
A new computational study adds to mounting evidence that electrophilic aromatic bromination reactions follow an addition–elimination pathway and not the classic mechanism described in textbooks.1
Brominating aromatic substrates is an important step in synthesising various pharmaceuticals, pesticides, flame retardants and dyes. Undergraduate organic chemistry courses often teach that such electrophilic aromatic substitution reactions proceed via a carbocation, known as a Wheland intermediate, that is conjugated but not aromatic. However, recent studies of these long-used reactions suggest they actually occur via direct substitution and addition–elimination pathways. For example, a DFT study in 2011 observed that the barriers to adding bromine to arenes in tetrachloromethane were smaller than those for direct substitution.2 Similarly, a joint computational and experimental study in 2014 found that chlorinating anisole proceeded via an addition–elimination pathway.3
Now, Frank De Proft from the Free University of Brussels, Belgium, and co-workers have used DFT to investigate the electrophilic bromination of benzene, anisole and nitrobenzene in both the gas and solution phases. They found that all three reactants favoured an addition–elimination bromination pathway. Moreover, they did not observe a Wheland intermediate in in acetonitrile, tetrachloromethane , or in the gas phase.
‘In contrast to what is often believed, this [study] shows that the kinetic control of the electrophilic aromatic bromination is not solely driven by the temporary loss of aromaticity upon electrophilic attack, but also by the strength of the electron delocalisation as well as a balance between attractive and repulsive non-covalent interactions between the reaction partners,’ notes De Proft.
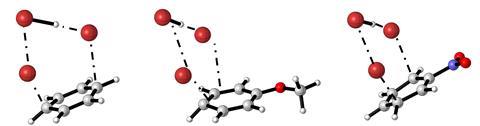
The researchers also investigated the reactions’ regioselectivity using a series of geometrical and electronic indices. Anisole was brominated preferentially at the para position due to stronger electron delocalisation and attractive interactions in the corresponding transition state. In contrast, the meta position was preferred for nitrobenzene due to steric clashes at the ipso carbon for the pathways leading to the ortho and para products. ‘For [arenes with electron-withdrawing groups], the rate-determining transition states … are chemically more like the reaction intermediate than the initial reactant,’ adds De Proft.
‘One thing I found amazing is that after 150 years of this reaction being reported, we are still discussing its reaction mechanism,’ comments Miquel Solà i Puig, an expert in computational chemistry and aromaticity from the University of Girona, Spain. ‘I think it is very important when discussing the reaction mechanism of this reaction to tell the students that the Wheland intermediate is a hypothesis that has been proven to be not correct.’
References
1 X Deraet et al, Phys. Chem. Chem. Phys., 2023, DOI: 10.1039/d3cp03137c
2 J Kong et al, Angew. Chem., Int. Ed., 2011, 50, 6809 (DOI: 10.1002/anie.201101852)
3 B Galabov et al, Proc. Natl. Acad. Sci. USA, 2014, 111, 10067 (DOI: 10.1073/pnas.1405065111)














No comments yet