New molecule-based method makes it easier to assess the precision of reactivity calculations
Swiss scientists have devised a method to warn chemists when their calculations of a molecular system’s reactivity should be treated cautiously.1 ‘To be truly predictive, we need to know where theoretical methods yield results affected by large errors,’ says Markus Reiher from ETH Zurich, the Swiss Federal Institute of Technology. ‘This is what our approach is now capable of.’ Using their method, Reiher and his PhD student Gregor Simm attached error bars to density functional theory (DFT) calculations for an entire catalytic nitrogen-to-ammonia conversion cycle.
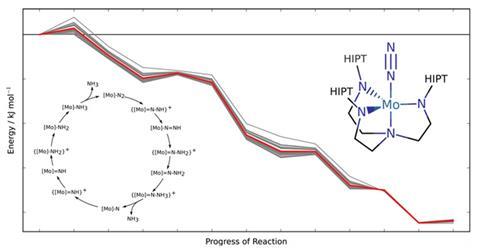
To calculate potential energies that determine reactivity, most chemists use DFT, which is quick, but often inaccurate. Error ranges for assessing conclusions based on DFT have been hard to get, Reiher explains. ‘We need error bars on observables that we calculate within a specific approach, rather than from comparing to reference data of utmost accuracy,’ he says. ‘Such accurate data are usually not available.’
Reiher highlights that other researchers had developed energy density functionals, the approximations at the heart of DFT, to produce error bars for solid state physics.2 However, that method didn’t transfer well from benchmark chemical systems to the ones scientists actually studied using DFT. This lack of transferability is particularly problematic for the transition metal complexes that interest Reiher and Simm.
In their new approach, the pair make a statistical link between two information sets. The first is a comparison of DFT estimates and reference data. The second compares the best result from an optimised density functional against the results the rest of the functionals of this class produce to get a confidence interval. Linking the two sets produces data scatter related to real errors when reference measurements aren’t available.
The Zurich team optimised their functionals’ parameters to be especially applicable to molecules with a metal centre. While this limits the applicability of their re-optimised functionals, it increases the transferability to similar molecules and allows for meaningful error assessment, Reiher says. And while this means all chemical systems need focused parameterisations, they are now trying to develop a more general method.
Aron Cohen, who works on improving DFT functionals at the University of Cambridge, UK, points out that commonly used functionals ‘still have large systematic errors’, particularly in catalysis. He calls the new approach ‘interesting’, but emphasises the trade-off inherent in sacrificing general applicability. ‘One has to apply extra effort but in return you have increased confidence in your results,’ Cohen says.
References
1 G N Simm and M Reiher, J. Chem. Theory Comput., 2016, DOI: 10.1021/acs.jctc.6b00318
2 J J Mortensen et al, Phys. Rev. Lett., 2005, DOI: 10.1103/PhysRevLett.95.216401










No comments yet