A case of mistaken identity inspires a closer look at the structural stability of Togni reagent I
Computational studies have unpicked the surprising stability behind high-energy fluorinating reagent Togni reagent I.1
Togni reagents – named after creator Antonio Togni – are trifluoromethylating agents that introduce the CF3 group often found in pharmaceuticals and agrochemicals. They’re members of a family of benziodoxole-based hypervalent iodine reagents that transfer atoms or functional groups loaded onto their oxygen and hypervalent iodine-containing five-membered ring.
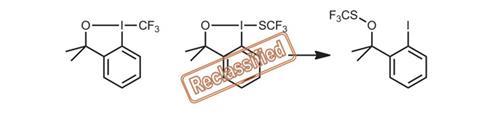
Stephen Buchwald and colleagues at Massachusetts Institute of Technology, US, recently called the structure of a new SCF3-substituted addition to the benziodoxole-based hypervalent iodine family into question.2 Spectroscopic analyses, derivatisation experiments and the crystalline sponge method helped them reclassify the structure as the isomeric thioperoxide.
Inspired by this case of mistaken identity, Henry Schaefer’s team, at the University of Georgia, US, set out to rationalise this difference, and discover whether other family members might exist in the ether form. Using density functional theory (DFT), the group calculated the free energy difference between isomers for a series of benziodoxole-based hypervalent iodine reagents. In most cases, the five-membered isomer was thermodynamically favoured, whereas the trifluoromethylthiolating reagent redefined by Buchwald preferred the more stable thioperoxide form – consistent with experimental data.
Yet the bench-stable hypervalent iodine form of Togni Reagent I was much higher in energy than the ether. ‘The five-membered ring ought to fall apart with the energy it has, yet somehow it doesn’t,’ Schaefer tells Chemistry World. So how can such a highly-strung compound be available to buy in a bottle?
After ruling out homolytic, heterolytic and bimolecular decomposition pathways, Schaefer’s team modelled transition states for plausible reductive elimination pathways from the hypervalent iodine form to the ether. DFT calculations found the transition state for isomeric conversion for the Togni Reagent too high to reach at room temperature. Schaefer says it may stem from favourable interactions between the orbitals on the iodine and p-orbitals on the benzene ring, or steric confinement from oxygen’s lone pairs.
Computational chemist Hans Peter Lüthi, from the Swiss Federal Institute of Technology in Zurich, notes how important theoretical studies are for understanding experimental observations: ‘The work is an excellent illustration of the usefulness of quantum mechanical modelling – it’s very interesting work and will possibly open the door to new hypervalent iodine chemistry’.
References
TS Sun et al, Chem. Commun., 2016, DOI: 10.1039/c6cc00384b This article is free to access until 21 April 2016
EV Vinogradova et al, Angew. Chem. Int. Ed., 2014, 53, 3125 (DOI: 10.1002/anie.201310897)









No comments yet