Deep learning algorithms set to transform time-consuming molecular screening programs
‘It’s an art and a science,’ explains Joshua Staker, a senior scientist at the US software company Schrodinger. He’s referring to deep learning – a branch of computer science that looks set to transform how chemists screen molecules and explore chemical behaviour.
Over the past few decades, deep learning has entered the public consciousness through projects such as AlphaGo. A landmark in computing, Google’s algorithm is able to autonomously learn and play the board game Go – 1050 times more complex than chess – a challenge once thought to be beyond computers. AlphaGo first defeated a human opponent in 2015, and beat the world number 1 in 2017.
Using algorithms to play games may seem of limited use in science. But if a machine can learn the rules of a game by playing itself, it can learn the rules of chemistry just by analysing chemical data. Deep learning platforms can quickly develop a knowledge of chemistry without any human instruction, and chemists are starting to realise that knowledge can be a powerful tool.
The beauty and simplicity of it is that there are no rules or features we need to engineer
Joshua Staker, Schrodinger
In Schrodinger’s case, Staker and his colleague Kyle Marshall wanted to speed up the process of screening for new drugs and materials by using deep learning to scour the literature for candidate molecules.
Research papers and patents contain huge numbers of molecular structures and experimental data that could be used in virtual screening programs, but getting it out of the documents is laborious. ‘First you have to identify what compounds in the publication you want to actually extract,’ comments Staker. ‘So, you read through the paper and then … go into some drawing software and draw it manually.’ Once the molecule is re-drawn in a computer-readable format (commonly known as SMILES), the information can be used in a screening program.
‘Doing this for hundreds of compounds in a large patent, it becomes tedious,’ laments Staker. ‘[It] starts to become easier and easier to make mistakes over data entry.’
Cutting out the middle man
Staker and Marshall came up with a solution to cut out the middle man. In fact, to cut out men and women altogether. The team has developed a deep neural network that can find images of molecular structures in a document and convert them into a digital format, without being told anything about molecules beforehand.1 ‘That’s really the beauty and simplicity of it in that there are no complex rules or features that we need to engineer as humans,’ says Staker.
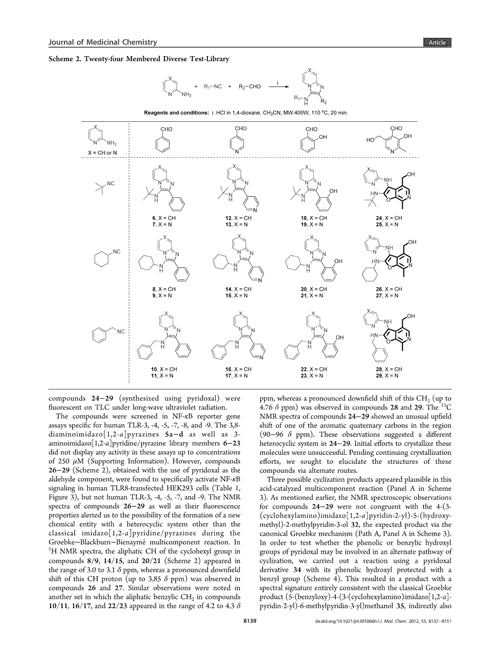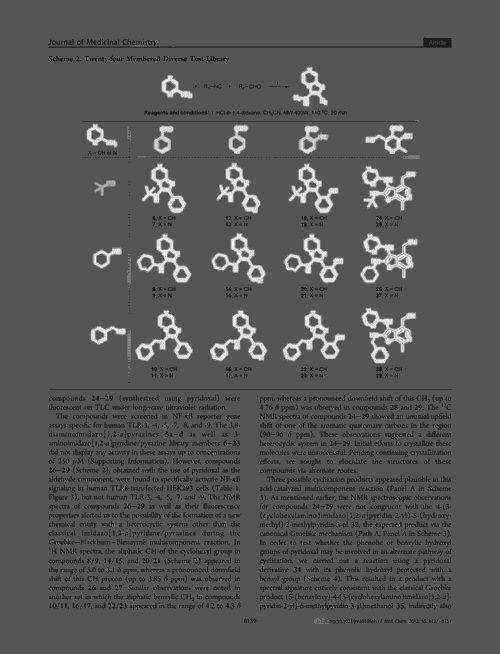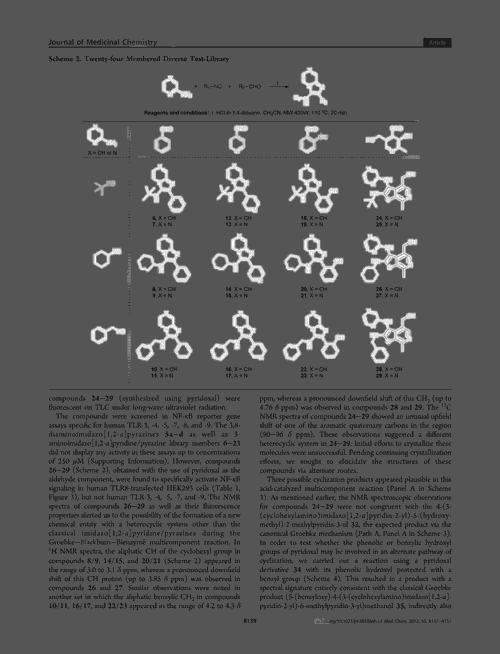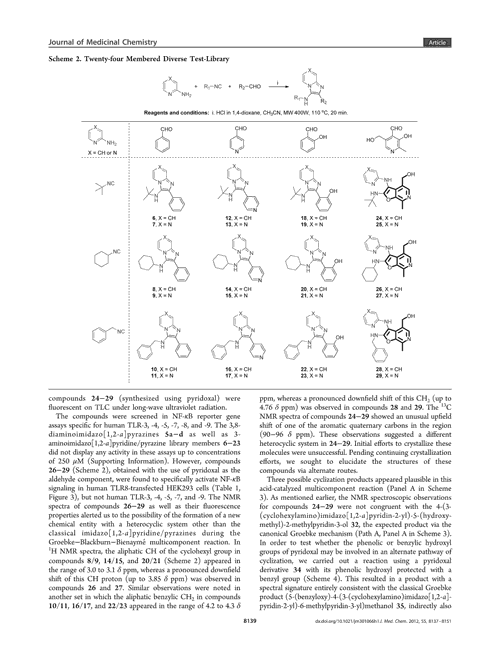
The platform comprises two separate networks. ‘We have a model that will recognise in an image what is and what is not a molecular structure – identifying the bonds, the atoms and what not,’ Staker explains. The software then cleans the image, separating it from nearby text, graphs or even other molecules. Marshall likens this segmentation process to how an image is cropped in photography software. The network can even recognise blurred or low-resolution molecules.
The second network then converts this image into a SMILES format using an encoder and a decoder. The image is first encoded as a series of co-ordinates, before a decoder interprets this vector as a SMILES string.
Mirroring intuition
To prepare the algorithm, the team trained it using sets of structures and their corresponding SMILES. Staker estimates the software saw around 128 million image–SMILES pairs before it was ready to transform blurred images into clean SMILES. Testing the algorithm on a series of low-resolution molecular libraries, the platform managed to identify up to 83% of the samples it was given.
It’s going to change the field, I’ve no doubt
Alán Aspuru-Guzik, Harvard University, US
After its extensive training, the algorithm has even adopted a seemingly human trait in some rare instances – intuition. ‘It’s seen so many chemical structures that it has learned a bit of chemistry,’ comments Staker. ‘If you were to put in something that wasn’t chemically accurate … it does have a chance to correct for that.’
The pair go on to explain how the algorithm, in some cases, can correctly assign chirality to atoms if it’s been missed in the original document. ‘It was as if the model was learning some intuition about what chirality means,’ posits Staker.
But the network is not intended to serve as a chemical savant – it is primarily designed to simplify the process of getting structures out of documents and into computers; offering researchers a well-curated library of molecules to use to find new drugs or new materials.
Perceiving predictions
However, other groups are building frameworks specifically designed to predict a molecule’s properties from its structure. This is the driving force behind Chemception, a framework developed by Garrett Goh and his colleagues at Pacific Northwest National Laboratory (PNNL) in the US.2 Given a molecule input, Chemception uses everything it has learned about how structures are related to properties to predict how that molecule will behave.
The group started with a challenge: what is the minimum amount of chemical knowledge an algorithm needs to predict a particular property? Nathan Baker, Goh’s collaborator and director of PNNL’s advanced computing division, likens such a network to dealing with an ‘untrained infant’. As with Schrodinger’s work, the infant goes about learning by first being trained on a dataset: a group of molecules labelled with data on their toxicity, anti-HIV activity and solvation energy.
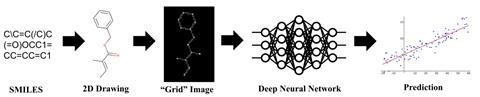
‘Instead of putting [Chemception] through a condensed chemistry curriculum … we drown it in labelled data and ask what it extracts from it,’ explains Baker.
Rather than starting with an image, the process begins with a SMILES description. This is converted into a 2D drawing which is fed into the Chemception neural network. Using the raw image alone, the platform then determines the molecule’s potential toxicity, activity or solubility. The team compared Chemception’s performance with networks specifically engineered for these tasks. In most cases, Chemception was able to match the accuracy of the specialised networks.
Tools such as quantitative structure–activity relationship (QSAR) models already perform this function. But such models have been built using decades of chemical knowledge, explains Baker, with humans providing the rules. ‘A lot of the progress in QSAR has been based on the development of better feature sets to describe molecules,’ he continues. Chemception, on the other hand, builds its own model from scratch. ‘In this case, it was just [using] 2D pictures of molecules,’ Baker tells Chemistry World.
We drown [the network] in labelled data and ask what it has extracted
Nathan Baker, Pacific Northwest National Laboratory, US
However, even though Chemception works with no prior knowledge, Baker is quick to point out that QSAR models do a lot more than simply predict properties. They can also enrich our understanding of why molecules have such properties in the first place – and that’s where he sees machine learning going next.
For Baker, a deep learning tool that can replicate QSAR’s ability may pay dividends in the biophysics community, where there is a need to understand amino acid behaviour. In particular, deep learning may help to reveal how pH affects protein interactions in their cellular environments.
Electric dreams
These potential applications are all rooted in a system that classifies molecules – also known as discriminative models. But could you create an AI that can generate new molecules, tailor-made to your specifications? Alán Aspuru-Guzik, a computational chemist from Harvard University, US, and his team have made it their mission to find out.
To explain their thinking, Aspuru-Guzik turns to science fiction: ‘Have you watched Blade runner?’ he asks. Based on the novel Do androids dream of electric sheep? by Philip K Dick, Blade runner tells the story of Deckard, who is tasked with hunting down synthetic humans (or androids).
‘Deckard … he’s supposed to be identifying androids; they could be androids, or they could be human,’ explains Aspuru-Guzik. This is the principle behind discriminative models. ‘In the chemistry community, it’s basically the idea of given an x, predict a y … my group and I used to use that to screen for molecules,’ he explains. But the androids themselves are generative models, Aspuru-Guzik continues. ‘They have to actually generate human behaviour and pass as a human.’ Along with his team, Aspuru-Guzik has now created a model that can both explore chemical space and essentially generate molecules with ideal properties.3
Like Chemception’s infant, Aspuru-Guzik’s algorithm starts with SMILES strings. But unlike Chemception, this infant is ‘unsupervised’ – there is no training set of worked examples, so the computer is left to find relationships in the data by itself . The team used a form of AI called an autoencoder, which receives and compresses information, be it in the form an image, document or even the film Blade runner, and then maps it in a ‘latent space’ by grouping similar data points together. Once mapped, the AI can explore this space and produce an estimation of the original input. Aspuru-Guzik likens it to how our brain constructs dreams.
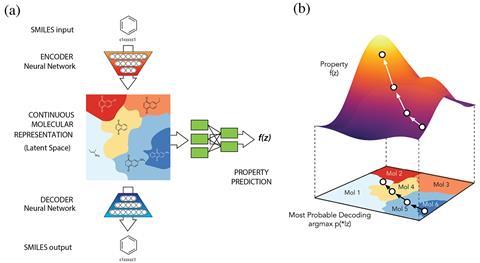
The team’s auto-encoder received up to 250,000 drug-like molecules in a SMILES format and mapped them to create its chemical latent space. But rather than simply generate an estimation of the original input, the team added another neural network.
‘You can add a third neural network connected to that sub-space that correlates to the property of the molecule,’ says Aspuru-Guzik. The AI can then explore this space with the goal of selecting a molecule with a desired property, such as drug-likeness or synthetic accessibility.
The model can also provide a user with all of the molecules that bear a resemblance to a known drug. ‘We took caffeine and we took aspirin and we said: “Give me the molecules between caffeine and aspirin”,’ comments Aspuru-Guzik. ‘As we move along in that line, we can decode and see what molecules there are – you can see the molecule morph into the other.’
Practice makes perfect
Deep learning’s capabilities are impressive, but it won’t be a replacement for existing computational tools, or even humans. Baker hopes deep learning platforms will complement current analytical methods to support chemists, not supplant them: ‘I would like machine learning to get to a point where it’s a tool in our toolkit alongside traditional molecular simulation, where not only does it help us generate answers … but we can tie it back to the underlying chemistry and physics.’
To achieve this, it may well take human ingenuity to design more robust, intuitive deep learning algorithms. Staker likens these design efforts to Lego building, where we can see both the art and science in building creative structures. But without a simple instruction manual, constructing such complex networks still requires one thing, according to Staker: ‘Really, it comes down to practice.’
For Aspuru-Guzik, the future of dream-like deep learning in chemistry is clear. ‘It’s going to change the field, I’ve no doubt,’ he says. ‘People that don’t see it, well let them wait.’
References
1 J Staker et al, 2017, arXiv: 1802.04903
2 G B Goh et al, 2017, arXiv: 1706.06689
3 R Gomez-Bombarelli et al, 2017, ACS Cent Sci, 2018, 4, 268 (DOI: 10.1021/acscentsci.7b00572)











No comments yet