
An alternative use for skeletal editing provides a one-pot method to synthesise ‘matching pairs’ of pharmaceutical candidates for structure-activity relationship (SAR) studies. This functional group transposition employs a photochemical reaction to exchange two ring carbons in dihydrobenzofuran substrates, simultaneously switching the position of any associated functional groups to produce the constitutional isomer.

Dihydrobenzofurans are a common motif in many bioactive compounds and derivatives and isomers of these structures are therefore important candidates in discovery chemistry. Compounds which differ in the position of the functional groups, known as constitutional isomers, are of particular interest. But, despite their similarity, these structures typically require independent syntheses, duplicating the effort and investment required to investigate them. Methods to directly transpose functionality are almost exclusively limited to highly unsaturated systems and, consequently, migrating substituents at a late stage is often a complex and lengthy procedure.
However, approaching this challenge from a different angle, Richmond Sarpong and his team at the University of California, Berkeley harnessed the power of skeletal editing to tackle this troublesome isomerisation in a simple one-pot reaction. ‘We imagined a scenario where a peripheral substituent could be formally moved to an adjacent position by breaking and reforming bonds at the core of the molecule,’ Sarpong explains.
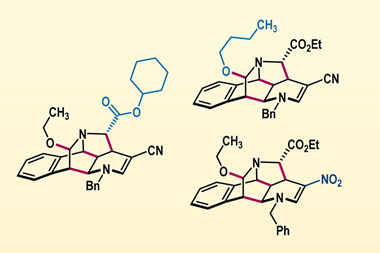
The team employed a photochemical reaction to isomerise acyl-substituted dihydrobenzofurans into a spiro-cyclopropane intermediate which they trapped with dilute hydrochloric acid. Subsequent elimination of this halide under basic conditions then facilitated a cyclisation to reform the benzofuran ring with the C2 and C3 carbons – and their carbonyl functionality – transposed. Simultaneously, they also developed a parallel neutral pathway, using a metal halide salt to alter the entire transformation in a single step.
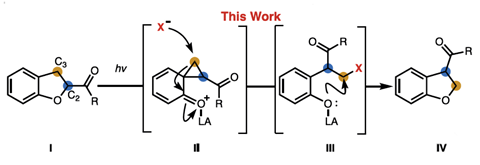
With these two sets of conditions established, the group investigated the scope of this unusual reaction. The acidic pathway effectively tolerated both electron-donating and -withdrawing substituents while the neutral conditions favoured substrates bearing basic groups. Acyls, esters, amides and carboxylic acids were likewise readily transposed under acidic conditions and the team even demonstrated successful transposition in two candidates from recent SAR campaigns.
‘We found, empirically, that these two sets of conditions were complementary,’ says Sarpong. ‘We do not fully understand the trends (which are still emerging) but it appears that in certain cases, the electronics of different portions of the substrate molecules makes them better behaved with one set of conditions over the other.’ The team are continuing to investigate these reactivity trends and hope to develop a deeper understanding of the underlying mechanism to extend the reaction towards other pharmaceutically relevant heterocycles such as indolines.
Bill Morandi, an organic chemist at ETH Zürich, believes this work addresses an important gap in our current synthetic capabilities and is intrigued to see how Sarpong’s methodology could be leveraged for the late-stage derivatisation of drug candidates. ‘Functional group transpositions are highly underdeveloped but I think they could have a transformative impact on molecular editing, particularly when able to transpose polar groups [like] alcohols, amines [and] acyls,’ he says. ‘The reaction developed by Sarpong is very creative and is poised to provide new avenues to important compounds and inspire the community at large to push for developing many such reactions for different functional groups.’
References
RT Steele et al, Science, 2025, DOI: 10.1126/science.adv9915











No comments yet