(+)-Heilonine
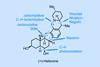
A short route that brings together the classical and cutting-edge
I’ve never tried ChatGPT. I’m not sure if this will be surprising to the readership of this column, but it certainly makes me a standout among my coworkers who use it constantly for anything from checking grammar and changing the tone of emails to writing rap battles between drugs and composing haikus on process development. Now I’m no luddite, and I’m not against the technology per se, but I’m not quite sure where it adds value or would fit into my day-to-day. Unlike most of my co-workers, I also enjoy the process of writing.
I’m sure this gap between invention and adoption can be seen in many fields, and it’s certainly present in organic chemistry. Even the most widely-read and knowledgeable bench chemists can be leery of new reactions and technologies – and I am as well. The thing is, even if we believe they’ll work, is it really worth the trouble of buying new catalysts or equipment? It’s hard to keep on top of the pros and cons of all the new methods being published, so why not stick with the safe route, even if it’s a step or two longer?