A total synthesis that uses a cunning trick to negotiate nitrogen
Alkaloid chemistry is no picnic. In fact, when I started my graduate studies, it was often joked that every nitrogen in your natural product would add a year to the time it’d take you to finish your PhD. Ever the empiricist, I found this to be true.
You see, even with careful planning, nitrogen-containing molecules will inevitably throw up roadblocks that’ll send you on lengthy detours or desperately hunting for workarounds. Unprotected amines alone are a challenge – to say nothing of the legendary intractability of guanidines. For one thing, they’re among nature’s strongest bases and nucleophiles. In addition, they make purification by conventional chromatography all but impossible – yes, I’ve tried adding triethylamine! To sidestep these issues, even virtuosic organic chemists will swaddle them with protecting groups to blunt their reactivity but, even so larded, they can still cause havoc with their lone pairs.
So why bother? Well, the very characteristics that make nitrogen a liability during multi-step synthesis also endow it with the ability to form strong interactions with proteins – and thus, bioactivity. In fact, a 2014 analysis found that 84% of FDA-approved drugs contained at least one nitrogen atom.1 The ubiquity of nitrogen in drug-like molecules is one of the reasons that catalysis in the pharmaceutical industry is often such a struggle; there’s usually a good reason that hot new methodology has no basic nitrogen atoms in the substrate scope! I worked with a nitrogen-rich drug candidate a few years back that could pull the iron out of reactor walls and bound palladium so tightly that you could column the complexes. Fortunately, we never had to make it on commercial scale.
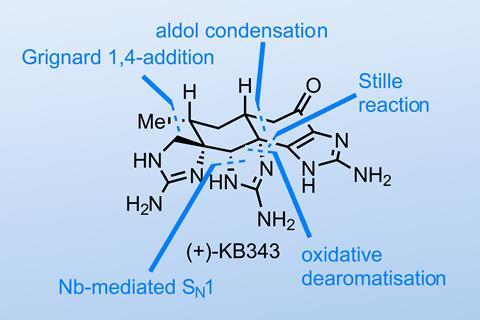
The guanidinium toxin KB343 is a redoubtable-looking molecule containing nine nitrogen atoms arranged in three cyclic guanidines, but a recent synthesis from the Baran group at Scripps Research in California, US, manages to make short work of it.2 That said, their creativity and hard work are apparent in almost every step, with a slew of unusual conditions and transformations that show just how many hundreds (if not thousands) of reactions they had to run to find a path through this nitrogen-laden minefield.
The team reports both racemic and enantioselective routes to the target but as a Diels–Alder afficionado, I can’t help but show the asymmetric synthesis to the molecule’s cyclohexanone ring (figure 1). It’s pretty safe to say that any given cyclohexanone can be made via Diels–Alder; the better question is how many steps will be needed as only certain diene/dienophile combinations work well. Here, the team carries out an unusual variant using two nitrogen-containing partners and a cobalt catalyst to get to the desired building block in short order.

Next, we pick up the synthesis with the team trying to convert a benzylic alcohol to an amine – a seemingly reasonable transformation with a dozen or more reliable options, none of which work in this case. In fact, even converting the alcohol to a leaving group proved impossible. The team was able to generate the quinone methide, but the only nucleophile they could add was hydroxide. After a lot of screening, the team was able to displace the alcohol with azide under SN1 conditions but obtained a single diastereomer resulting from retention – the wrong one. This could be reduced to the amine with stinky, pyrophoric trimethylphosphine but to get it to cyclise the team had to invert the stereochemistry through oxidation and reduction (figure 2). This is a common trick for alcohol inversion, but it’s far from trivial for nitrogen – I’ve never seen this executed in a total synthesis before!

Another thing that’s hard to convey here is how cleverly the team walk the guanidines around the molecule to minimise protecting group chemistry and selectivity issues. I’d definitely recommend the full paper as a masterclass in modern alkaloid chemistry.
References
1 J T Njardarson et al, J. Med. Chem., 2014, 57, 10257 (DOI: 10.1021/jm501100b)
2 P S Baran et al, J. Am. Chem. Soc., 2023, 145, 7753 (DOI: 10.1021/jacs.3c01991)









No comments yet