Choosing an efficient route to a classic target
At my industry day job, I’ve often seen a process chemistry team struggle to pick a route to a drug candidate. While they might have five objectively good routes to choose from, they must ultimately pick just one and it often seems that ‘best’ is in the eye of the beholder. You see, commercial route selection is a multiple variable problem and it’s not uncommon to see teams pass over a shorter route in place of an operationally simpler one, or a cheaper one in favour of a safer one.
In academia, the selection pressures are certainly different, but the aim is still to end up with the best route – whatever that means. Aphoristically, it’s often said that comparison is the thief of joy, but I’ve always enjoyed contrasting the different routes that research groups devise to conquer a natural product. Popular targets with a large number of reported syntheses are a unique learning opportunity as we get to see how different chemists attack the same problem.
Tetrodotoxin is a classic but still highly challenging target, looking almost as intimidating today as it did when Yoshito Kishi first synthesised it some 50 years ago. Since then, another 11 syntheses have been published, as brave chemists test their intellect and hottest new methodologies against this formidable target. Here, I’m going to be discussing the recent approach from Dirk Trauner, who moved to the University of Pennsylvania, US, this summer, and co-workers.1
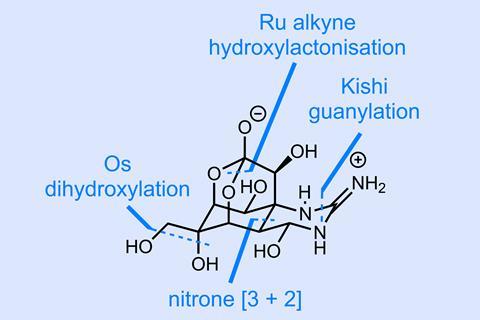
The vast majority of tetrodotoxin syntheses reported to date either buy the molecule’s core carbocyclic ring, or build it early on in the synthesis. In either case, this usually requires a lot of subsequent legwork to then install the huge number of heteroatoms present in the target. Trauner holds off on closing this ring until later in the synthesis, instead taking seven steps to shape a glucose-derived starting material into a nitroalkene. This intermediate seems a long way off from the target – consider that we’re almost a third of the way through the synthesis at this point – but with an interesting dipolar cycloaddition the synthesis suddenly surges forward. To kick off this transformation, p-methoxybenzyl alcohol (PMBOH) is deprotonated and added into the nitroalkene; treatment of the resulting nitronate with di-tert-butyl dicarbonate (Boc2O) then generates a nitrile oxide that cyclises to give the product in excellent yield and with near-perfect stereoselectivity (figure 1). It should be noted this is a particularly effective way of setting the tricky C-8 stereocentre, which can now direct acetylide addition to the imine, although this comes at the cost of the incorrect (albeit easily corrigible) configuration at the C-4a stereocentre.

Fast forward past alkynylation, N–O bond cleavage and a handful of non-strategic operations (nothing to see here!) and the team is ready to close the lactone ring. After – I suspect based on the non-obvious reaction conditions – a significant amount of tinkering, the team arrives at a really elegant and unique solution. First, the alkyne is treated with a ruthenium catalyst, rendering the terminal carbon electrophilic via a metallo-vinylidene carbene and effecting cyclisation to the dihydropyran. Oxone is then added in the same pot, converting the ruthenium cycloisomerisation catalyst into an oxidant, which cleanly takes the dihydropyran to the hydroxylactone (figure 2).

From here, there’s just the installation of the guanidine group, deprotection and cyclisation to get to the target. Although not quite the shortest to date, this elegant synthesis is shockingly efficient with an average yield of almost 90% per step – very impressive for such a dense and highly oxygenated target. Congratulations to the team on an impressive addition to the tetrodotoxin canon!
References
1 D B Konrad et al, Science, 2022, 377, 411 (DOI: 10.1126/science.abn0571)









No comments yet