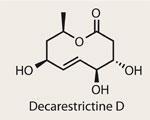
Decarestrictine D
Although its chemistry is mature and varied, my use of silicon reagents in my synthetic forays has been limited to a somewhat clumsy use of hydroxyl protecting groups. Worse still, I have neglected its group 14 colleague, tin, even further - only using stannanes in the ubiquitous Stille coupling. This now feels like an opportunity lost, as a team lead by Varinder Aggarwal of the University of Bristol, UK, demonstrates their synthetic utility beautifully in a short formal synthesis of the cholesterol biosynthesis inhibitor, decarestrictine D.1 With four stereocentres clustered around an unsaturated bond, this small but complex lactone is the perfect challenge to showcase a powerful method.
β-hydroxy allylsilanes have long been used as powerful reagents in organic chemistry, but their synthesis can be quite awkward. This problem is only enhanced when the desired allylsilane is chiral - a problem the group set out to solve. Their plan was to combine allylsilane synthesis with a boron-mediated aldehyde addition to produce a novel three-component coupling - an ambitious goal, but very appealing.
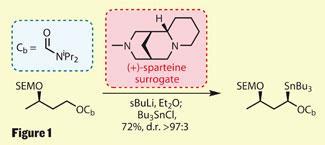
Their synthesis started with construction of a chiral building block (figure 1), containing one silyl-protected secondary alcohol and a carbamate-protected primary alcohol (denoted OCb). Treating this with a strong base - in the presence of a chiral diamine to direct the deprotonation - allowed the team to form a chiral carbon-tin bond in high diastereomeric excess. In this case, the source of chirality was a ’(+)-sparteine surrogate’. (-)-Sparteine is an alkaloid found in plants of the lupin family and a very useful chiral reagent, but the (+)-enantiomer is not easily available. Thanks to this recently developed substitute,2 both options are now open to chemists.
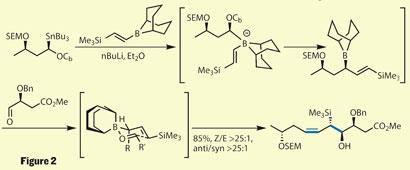
With their chiral stannane fragment complete, they moved on to a pair of coupling reactions to build up much of the target’s complexity. Exchanging the tin for lithium gave an asymmetric carbanion, which attacked the vinyl borane and rearranged to substitute the carbamate for the vinyl silane group. Addition of a small aldehyde fragment allowed formation of a cyclic chair-type transition state (figure 2). As many readers will remember from undergraduate organic chemistry, this type of transition state often gives excellent stereoselectivity, and after a little tinkering this proved to be no exception. The result of their efforts was the fluid installation of a cis double bond, along with the creation of a new stereocentre. The group performed this chemistry on a wide variety of substrates outside the scope of the total synthesis - indeed this is fundamentally a methodology paper - but I was quite taken with the rest of their synthesis.
An elegant ending
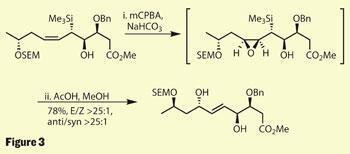
The team weren’t quite done with their manipulation of group 14 elements. Using some silicon chemistry developed decades earlier, they were rather cunningly able to use an acidic Peterson rearrangement to install both a hydroxyl stereocentre and create the desired trans double bond.3 This worked by first oxidising the cis olefin with meta-chloroperbenzoic acid (mCPBA), which was highly stereoselective - directed by the bulky silicon group. Treatment of the now fragile epoxide intermediate with a little acid resulted in a stereospecific elimination of the silicon group, and creation of the required trans alkene (figure 3).
From this point, the group could complete the target in five steps; but three steps was enough to consititute a formal synthesis (in which their route converged with a previous total synthesis). Importantly, this route is not only shorter than previous efforts, but also acts as the perfect test of a new and useful method. Perhaps I should brave the smell of rotting leaves (that’s what silicon reagents remind me of) more often; carbon shouldn’t be the only group 14 element in frequent use.
Paul Docherty is a medicinal chemist based in London, UK
References
- V K Aggarwal, M Binanzer and G Y Fang,Angew. Chem. Int. Ed. , 2010, DOI: 10.1002/anie201001223
- P O'Brien et al, J. Am. Chem. Soc., 2002, 124, 11870.
- W R Roush and P T Grover, Tetrahedron Lett., 1990, 31, 7567












No comments yet