Hypocrellin A
As a rule, Tot. Syn. has plenty of time for complex targets and mechanisms, but has an anxiety about ’Big Concepts’ - if it can’t be explained by curly arrows, all is lost. However, the latest synthesis by Marisa Kozlowski of the University of Pennsylvania combines all three in an appealing package, so it’s time to get the thinking caps on.1
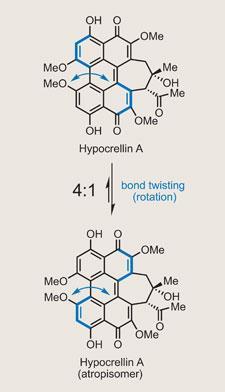
The target is hypocrellin A, which couldn’t look much less like last month’s callipeltoside A. Even a casual glance reveals one intriguing feature of this target - the fact it exists in equilibrium with an isomer. In this case, the type of isomerism at play is atropisomerism, where stereoisomers result from restricted rotation about a single bond. This is down to the non-planar nature of that polycyclic aromatic core - much like the well-known BINAP ligands.
This feature complicates both the NMR spectra of the target (mixtures of compounds always present a headache), and the synthesis. However, the group had a cunning strategy in place. In fact, so confident were they about controlling the helical chirality, they planned to use it to direct installation of the two more traditional stereocentres in a dynamic stereochemistry transfer reaction.
On seeing this, my thoughts were, ’a what?’ But clearly Kozlowski anticipated my confusion, and provides an explanation. The idea is to use a precursor, in which helical isomerisation is suppressed at room temperature, to control the stereochemistry of the key intramolecular aldol reaction. This twist in the molecule should direct formation of the two stereocentres as the final seven-membered ring is installed.
However, rather counterintuitively, introducing that last ring drops the thermodynamic barrier to atropisomerism to room-temperature or below. This starts the molecule flipping between the two stereoisomers - hence dynamic stereochemistry transfer.
Down to business
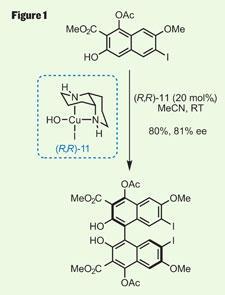
First challenge was to install the helical chirality - a Kozlowski group speciality. They’ve developed a relatively simple and effective method for this, using a chiral copper catalyst to provide stereocontrol (figure 1).2 This dimerisation of an easily accessible naphthol (produced in five steps from commercial starting material) provided the core of the natural product quickly and efficiently, and importantly in a good enantiomeric excess. This was boosted further to more than 99 per cent ee by trituration to simply wash away soluble impurities - nice to see an old-school technique in such a modern synthesis.
A few steps later, it was time to loose a couple of superfluous carboxylic acid groups in a decarboxylation. Although methods for this reaction are well known, all involve heating the hell out the starting material and praying for something other than treacle. This presents quite a problem, as the 180?C required is beyond the thermodynamic barrier to atropisomerism, resulting in racemisation. Their solution to this problem was to develop a new palladium-mediated process, 3 at the relatively cool 70?C, which provided the product in a reasonable yield. (The methodology work cited suggests that catalytic use of the palladium complex is effective, but they actually used 2.5 equivalents in this synthesis. ouch!)
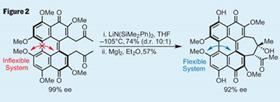
After a few functional group conversions, and removal of two ketal protecting groups, it was time to test that high-concept aldol reaction. Treating the 1,8-diketone with a bulky lithium base gave the desired seven-membered ring in good yield and diastereomeric ratio, with only a little leeching of enantiomeric excess, bringing the ’Big Concept’ to practice (figure 2).
The final reaction to complete the target was also quietly impressive - three different aryl methyl ether environments existed in the aldol product, but only two are present in the target. This means attempting a chemoselective demethylation as the last step - quite a risky strategy. However, a little magnesium iodide was quite effective, giving the target in a reasonable yield and completing an intriguing synthesis.
Paul Docherty is a DPhil student at the University of Oxford, UK
References
1 M C Kozlowski et al,Angew. Chem. Int. Ed., 2008, DOI:
10.1002/anie.200800734 2 M C Kozlowski et al,J. Org. Chem., 2003, 68, 5500 3 M C Kozlowski et al,Org. Lett., 2007, 9, 2441












No comments yet