Iriomoteolide
Iriomoteolide
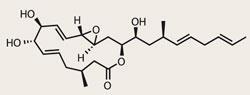
The hunt for anticancer therapies is inextricably linked with natural products (such as taxol), and more specifically with macrolides. Families such as the amphidinolides, produced by various sea dwelling Amphidinium microorganisms, have proven to be exceptionally active against cancer cell lines, and are currently in clinical trials. More recently, a new class of Amphidinium macrolide named the iriomoteolides were discovered, featuring a pretty special 15-member-ring macrocycle. That’s certainly enough to interest the keen synthesiser, but not enough for those pesky grant applications. Happily, a potent cytotoxicity against lymphoma cell lines was clearly enough to bring in the Swiss francs for Cristina Nevado of the University of Zurich, and allow her team to complete a first synthesis.1
As a field for the prospective total-synthesiser, macrolides are rather mature, with plenty of reliable chemistry in the laboratory toolkit (especially the large section marked ’aldol’). However, certain structural features are amenable to a shortcut - and as spotted by Nevado, iriomoteolide contains a rather handy symmetric portion in its far left. Their intention was to append this section using metathesis chemistry - but first they needed something to append it to.
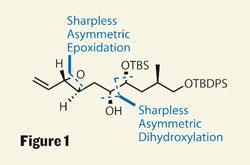
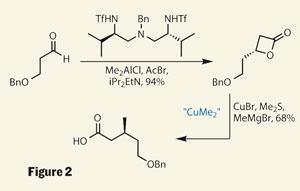
Four stereocentres were created using chemistry developed by Scripps’s Barry Sharpless, using his now famous (and Nobel prize winning) epoxidation and dihydroxylation reactions (figure 1).2,3 These dependable reactions allowed quick construction of the ’top’ of the target, with a more unusual approach then used for the ’bottom’. Using some recent work by Scott Nelson of the University of Pittsburgh, US, they were able to easily produce a chiral b -lactone.4 This sensitive intermediate was attacked by in-situ generated dimethyl cuprate, to add an isolated methyl group stereoselectively (figure 2).
Goldilocks effect
A simple esterification connected these two fragments, ready for the metathesis chemistry to begin. Also a Nobel prize winning reaction, this chemistry hasn’t quite reached the same level of predictability as Sharpless’s work - a common problem is that the organometallic catalyst can interact with any free hydroxyl groups in the substrate, causing side reactions nearby. In this case, Nevado was hoping to use a C2 symmetric substrate with two allylic alcohols (figure 3). This would, if the chemistry went perfectly, perform two metathesis reactions, the first a cross-metathesis to add the extra string of carbons, and the second closing up the 15-membered ring. However, a complex mixture of products was found, presumably due to the unprotected hydroxyl groups.
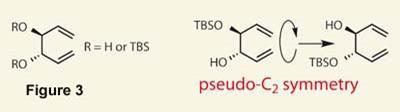
Protecting both hydroxyl groups as their TBS (tertiary-butyldimethylsilyl) ethers seemed to be the perfect method of reducing this overreactivity - but this went too far! The metathesis catalyst completely ignored the protected substrate, and instead caused the partner to form an undesired 10-membered ring.
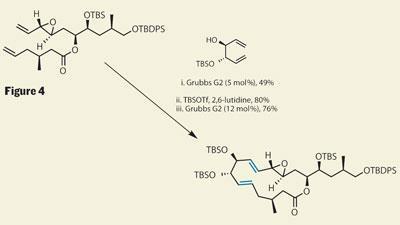
What the team needed was a third way - and one that is quite neat (and makes me think of childhood nursery stories...). Using a pseudo-C2 symmetric substrate, with one protected hydroxyl, turned out to be ’just right’, with a good yield of the cross-metathesis product achieved. A further protection of the remaining hydroxyl then allowed the key ring-closing metathesis reaction to complete, and completion of the macrocycle (figure 4).
A neat, selective deprotection of a primary silylether without affecting the less reactive secondary silylethers allowed the group to attach the final carbon atoms to the right hand side, and shortly afterwards, complete the target. However, before drafting this elegant paper, they decided to try and one-up nature by producing several analogues of iriomoteolide in the hope that one might be a more potent cancer-basher. Their small library of analogues didn’t turn up any such hits, but did suggest possible avenues for future research.
Paul Docherty is a medicinal chemist based in London, UK
References
1 R Cribiú, C Jäger and C Nevado, Angew. Chem. Int. Ed., 2009, DOI: 10.1002/anie.200903379
2 A Pfenninger, Synthesis, 1986, 89
3 H C Kolb, M S VanNieuwenhze and K B Sharpless, Chem. Rev., 1994, 94, 2483 (DOI: 10.1021/cr00032a009)
4 S G Nelson, T J Peelen and Z Wan, J. Am. Chem. Soc., 1999, 121, 9742 (DOI: 10.1021/ja992369y)












No comments yet