Stable Ni(IV) complexes could help nickel rival its flashier platinum-group cousins, says Karl Collins
Medieval German miners were vexed by an ore they named kupfernickel. It looked like a copper ore, but contained no copper. They blamed a mischievous demon (nickel) for replacing the copper (kupfer) they sought with the metal we now know simply as nickel – that nickel arsenide ore is now called nickeline.
In the world of transition metal catalysis, some still consider nickel less worthy than other metals – particularly palladium. But recent advances have increasingly brought it into the limelight.1
Some of the reasons nickel has increasingly become a favourite for contemporary catalysis include its unique physical characteristics; a particular aptitude in enabling reductive cross-couplings and functional group transformations; and the ability to undergo single-electron transfer and two-electron processes (for example oxidative addition and reductive elimination). Add to that facile oxidative addition and slow b-hydride elimination processes relative to palladium, and it is clear that nickel has great value in its own right.
Bringing nickel further to the forefront is a groundbreaking paper from Nicole Camasso and Melanie Sanford at the University of Michigan, US, which demonstrates the potential of Ni(iv) species in catalytic carbon–heteroatom bond formation.2 The chemistry of nickel typically exploits the metal in oxidation states from –1 to +3, although the door has now been opened to the chemistry of hitherto unexplored Ni(iv) species.
The team began with relatively simple cyclic voltametry studies of a bipyridyl Ni(ii) complex that suggested access to a Ni(iv) oxidation state might be possible. Initial attempts to identify a Ni(iv) species following oxidation with a variety of reagents common to Pd(ii)–Pd(iv) chemistry were unsuccessful, although the reactions do generate a cyclobutane product, most likely by reductive elimination of the C(sp2)–C(sp3) centres (figure 1).
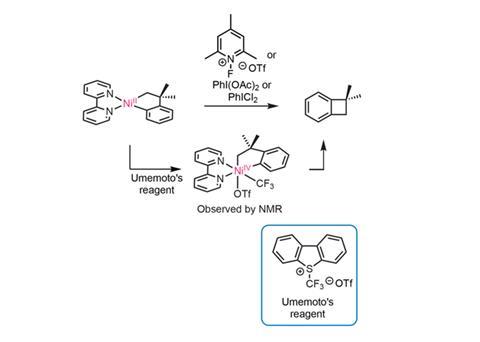
Working on the presumption that this reductive elimination was from a transient Ni(iv) species, the researchers sought a more stable complex to confirm the hypothesis. Switching the oxidant to Umemoto’s reagent (5-(trifluoromethyl)dibenzothiophenium triflate), a widely used trifluoromethylating reagent, produces a Ni(iv) complex stable enough to be characterised by NMR. Swapping ligands to tris(2-pyridyl)methane gives a Ni(iv) complex that persists in solution for several days and can be crystallised for x-ray analysis. This confirmed an octahedral configuration at the Ni(iv) centre.
Sanford and Camasso have explored the reactivity of this unusual complex, and show that it undergoes quantitative carbon–carbon reductive elimination to form the previously observed cyclobutane. More excitingly, on treatment with ammonium acetate, it selectively forms a C(sp3)–O bond at room temperature to give the acetate ester product complex in an isolated yield of 78% after 1.5h (figure 2).
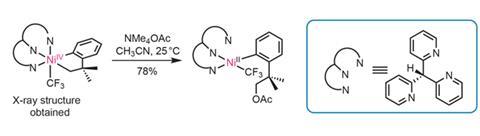
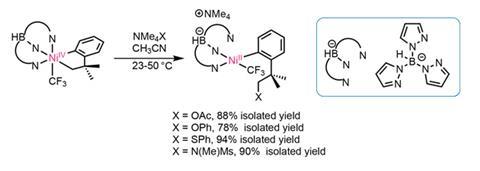
The team suspects that the highly electrophilic cationic nickel complex leads to unwanted side reactions with other heteroatom nucleophiles. Using the anionic tripyrazolylborane ligand instead forms a neutral Ni(iv) complex, which undergoes highly selective and high yielding C(sp3)–O, –N and –S bond formation (figure 3). This selectivity in bond formation may seem surprising when viewed as a reductive elimination pathway. However, mechanistic studies suggest an SN2 type nucleophilic substitution mechanism, which accounts for the selectivity. The team proposes that the nucleophile attacks the metal–carbon bond directly, similar to that seen in to Pd(iv) and Pt(iv) systems.
Sanford and Camasso stress that the job of exploiting Ni(ii)–Ni(iv) reaction systems in catalysis has only just begun. The first steps will be to identify ligands and oxidants that can selectively form Ni(iv) species, avoiding the pitfalls of single-electron transfer processes. While there is no doubt that this is a major challenge, Sanford’s expertise in mechanistic analysis and experience in high-valent metal catalysis suggest that her group is likely to lead the imminent charge on opening up this new frontier in nickel catalysis.
Karl Collins leads a medicinal chemistry laboratory in Germany











No comments yet