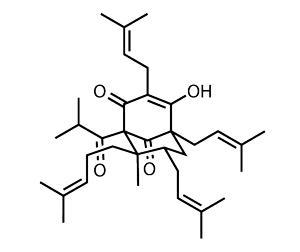
Studies of St John’s wort have found several biologically active ingredients, but the primary agent is hyperforin, a potent inhibitor of several neurotransmitters.1 Chemically, hyperforin is a polycyclic polyprenylated acylphloroglucinol – and typically for this family consists of a bicyclo[3.3.1]nonane decorated with several isoprenoid side chains. Whilst this structure confers potency to hyperforin, it brings with it considerable downsides – in particular low water solubility and high sensitivity to oxidation.
The challenge to medicinal chemistry is rather clear – provide analogues of hyperforin that maintain the efficacy of the natural product, but which are both more stable and water soluble. However, to do this, an efficient route to the target offering opportunities for selective and flexible derivation is required – something a team led by Matt Shair of Harvard University, US, has provided.2
Starting with the core of the problem, the US team worked towards two fragments that would deliver the complex bicyclo[3.3.1]nonane system. Derivation of geraniol provided the starting material and asymmetry, but rather early in the synthesis they used a reagent I’d rather have left on the shelf – mercuric acetate. Although nowhere near as toxic as other organomercury compounds, the by-products from oxymercuration reactions can be rather nasty, but the mildness of the protocol and utility of the reaction keep it relevant.
The second fragment also required a potentially toxic reagent in the form of barium iodide, but this was necessary to increase the selectivity of the prenylation reaction. Deprotonation of the skipped diene formed a pentadienyl anion, which could react with prenyl chloride at multiple sites, but using freshly prepared barium iodide focused the reaction at the desired location. A second deprotonation at the same centre facilitated smooth coupling with the first fragment, returning the cyclisation substrate directly.
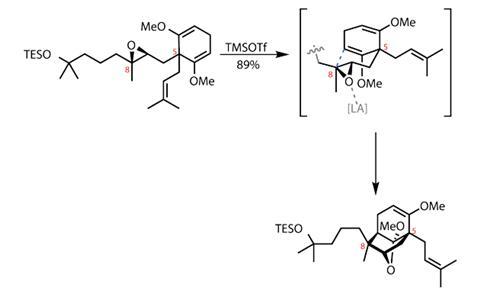
The cyclisation event itself was more straightforward – using a common Lewis acid to activate the epoxide prompted a rearrangement reaction, forming two rings in the process, with excellent stereoselectivity and overall reaction efficiency (figure 1).
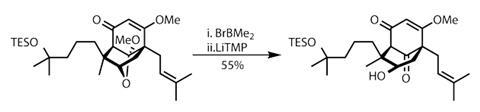
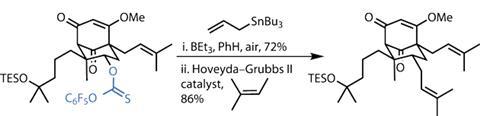
Paul Docherty is a science writer and blogger based in Reading, UK
References
(DOI: 10.1007/s002130000625)












No comments yet