
C–H activation takes the stress out of organometallic couplings
Organometallic conjugate additions made up a significant part of my PhD, and it was not a fun part. Cutting up lithium metal became a race against time as the dark grey slivers turned to white in the air. And transmetallation with deadly copper cyanide always made my heart beat a little faster, though nothing had more of an effect on my blood pressure than the cryostat that changed temperature whenever somebody walked past it. So I am always joyful to see β-functionalisations of carbonyl groups from outside the realm of classical organometallic chemistry and directed carbon–hydrogen bond activation has made some outstanding progress here.
Jin-Quan Yu continues to make outstanding contributions to the field of C–H activation
This approach inserts transition metals into carbon–hydrogen bonds, using nearby chelating groups to guide the metal into place. The subsequent reactive intermediates enable a multitude of different bond forming reactions from ‘unfunctionalised’ substrates. While the early flurry focused on the functionalisation of sp2 C–H bonds, the development of bulky bidendate directing groups opened up primary sp3 C–H bonds to directed activation. Nowadays, numerous directing groups are reported with clever designs allowing for meta-functionalisation of arenes or the highly challenging functionalisation of methylene sp3 C–H bonds. And with ‘transient’ directing groups installed and removed in situ and ‘traceless’ directing groups that vanish during the reaction, many critics of the field have at least partially been silenced. Yet truly utilitarian C–H bond activation reactions are still not as prevalent as we would like. Jin-Quan Yu of the Scripps Research Institute, US, has been working to change that.
In recent years, the Yu lab has delivered some of the field’s most outstanding papers, including developing an increasingly general β-arylation of ketones. In 2016, the team demonstrated a very limited proof of concept for palladium-catalysed arylation of methylene C–H bonds beta to a ketone, using glycine to form a transient directing group.1 This was followed up in 2017 by substituting the glycine for a β-amino acid, enabling formation of a six-membered palladacycle that proved more reactive and significantly extended the scope of the reaction, albeit largely limited to methylketones (figure 1).2
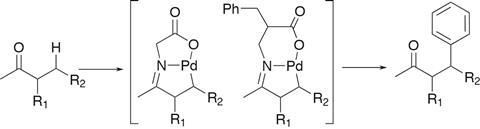
The most recent instalment arguably delivers a truly useful transformation, massively increasing reaction’s scope with respect to both the ketone and the (hetero)aryl coupling partner. But there is a proviso: in contrast to previous reactions, this work requires the pre-installation and subsequent removal of a directing group.3
The team began by installing two methyl groups on a known bidentate directing group – enhancing its co-ordinating ability by exploiting the Thorpe–Ingold effect. This enhanced the reactivity and gave the team a great starting point to explore the reaction of a whole host of ketones that were previously unreported, with a significantly increased tolerance to the steric and electronic environment of both reaction partners.
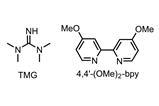
The team then used their new conditions in reactions with heteroaryl coupling partners. These partners often prove problematic in palladium mediated reactions because they compete with the directing group for coordinating the catalyst. Yet the functionalisation of cyclobutylketones with various iodopyridines proceeded in more than acceptable yields, giving access to products that simply cannot be prepared via conjugate addition chemistry (figure 2) – a very impressive outcome.
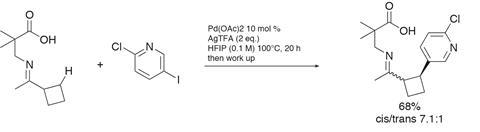
This is very important work from the Yu lab, though the details of this paper also highlight many negative aspects of the current publishing paradigm. The team writes of a ‘versatile’ reaction that addresses ‘all the limitations’ of previous β-C(sp3)–H arylations of ketones – which is not the case, and go so far as to describe these limitations as ‘fatal’. Many argue this kind of language is inappropriate in a scientific publication and often blame the authors, though editors and referees must also take responsibility. And the work is not without its own limitations: installing and removing the directing group require cooking the substrate in pyridine and 4N HCl at 100°C for several hours respectively, which would certainly prove fatal to even mildly sensitive substrates.
Jin-Quan Yu and his team continue to make outstanding contributions in the field of C–H activation, breaking the dogma of perceived conceptual limitations. This work is a great example of systematically addressing the real limitations of a given transformation. Though the journey is still not quite complete, I imagine many students will be happy to at least try getting rid of the cryostat and putting this reaction to the test. I certainly would have been!
References
1 F-L Zhang et al, Science, 2016, 351, 252 (DOI: 10.1126/science.aad7893)
2 K Hong, H Park and J-Q Yu, ACS Catal, 2017, 7, 6938 (DOI: 10.1021/acscatal.7b02905)
3 R-Y Zhu et al, JACS, 2017, 139, 16080 (DOI: 10.1021/jacs.7b09761)












No comments yet