A fully automated synthesis of fluorine-18 tracers suitable for positron emission tomography
Since the development of positron emission tomography (PET) in the 1970s, it has become an increasingly important imaging tool in biology. By detecting positron-emitting radioisotopes, PET can image the biological distribution of labelled drugs or biomolecules, allowing the study of disease and biological pathways. The technique can also determine if drugs reach tissues at effective therapeutic levels.
Carbon-11 and fluorine-18 are the most prevalent isotopes used as PET tracers. Synthetic chemical methods for carbon-11 are well established, but its relatively short half-life of 20 minutes means that radiotracers must be synthesised at the location they are going to be used. Unfortunately not everyone has their own cyclotron! On the other hand, fluorine-18 has a half-life of 110 minutes, which allows synthesised radiotracers to be transported, and permits more complex (ie longer) kinetic studies. Consequently, effective synthetic methods for fluorine-18 are increasingly sought after.
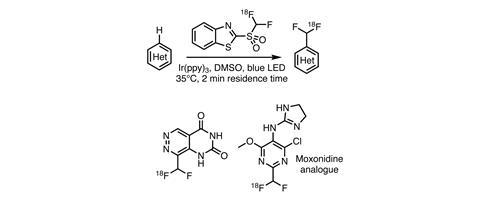
Fluorine chemistry has a well-deserved reputation of being the reserve of the most hardened synthetic chemists, since its methods are often harsh and dangerous. But recent advances in catalysis are providing safe and mild alternatives. Photoredox catalysis has played a role in enhancing the fluorination chemist’s toolbox. Last year, researchers at UCB Pharmaceuticals in Belgium, in collaboration with the GIGA-CRC in vivo imaging department at the University of Liège, Belgium, reported a mild photoredox method to introduce fluorine-18 labelled difluoromethyl groups directly onto electron-deficient heterocycles, including several drug molecules.1
Using what can be described as classical photoredox reaction conditions, the team generated a difluoromethyl radical from [18F]benzothiazole sulfone to functionalise electron-deficient heterocycles via a Minisci radical substitution reaction (figure 1). In a flow reactor, the model reaction gave the fluorine-18 labelled form of the antiherpetic drug acyclovir (Zovirax) in 70±7% radiochemical yield in just two minutes, with a molar activity suitable for clinical applications.
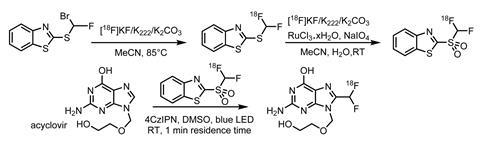
Unsurprisingly though, the development of radiotracers for human studies is more complex. High amounts of radiation and reproducible methods that meet stringent Good Manufacturing Practice standards mean fully automated syntheses are ideal, as the researchers demonstrate in their most recent work.2
Using an AllinOne synthesiser from Trasis, the team began by optimising the preparation of the difluoromethylating reagent. Starting with the bromine precursor, nucleophilic substitution with fluorine-18 labelled potassium fluoride gave the labelled benzothiazole in five minutes. This was then directly oxidised to give the reagent in a reasonable radiochemical yield of 10±1.6% and a high molar activity of 59.2±4GBq/µmol. The team then worked on automating the photochemical reaction, first developing a 3D-printed photoreactor compatible with the AllinOne synthesiser and then reoptimising the reaction conditions.
Initially, the light source in the new reactor generated a higher reaction temperature, which degraded the sulfone reagent, leading to a drop in radiochemical yield. Fortunately, exchanging the iridium-based photocatalyst for 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) restored the yield with the additional benefits of being cheaper and reportedly easier to remove from the reaction mixture.
The fully automated radiosynthesis performed using a high activity batch of [18F]fluorine (160GBq) gave the final compound in a total reaction time of 95 minutes, with a radiochemical yield of 1.4±0.1% and a molar activity of 35GBq/µmol – suitable for clinical PET studies (figure 2).
This is an excellent study that validates the fully automated photochemical synthesis of potential PET tracers with the sought-after safety and reproducibility aspects addressed, although there are limitations. Synthetically, Minisci reactions are often unselective and are not tunable when functionalising a specific molecule: you get what you get. Also, introducing a difluoromethyl group into a biologically active molecule may affect its behaviour in the body – in the worst case completely inhibiting its activity or leading to side effects. Finally, the radiochemical yield is low, though the team suggest that this arose from a compound-specific purification issue.
Some say that novel synthetic chemistry is of limited value in contemporary research. With synthesis increasingly being perceived as a service function in the life sciences, this belief is often reinforced in an industrial environment. While it may be true that synthesis is rarely a limiting factor in drug discovery research today, work such as this clearly demonstrates the impact great synthetic methodology can have to help us study disease.
References
1. L Trump et al, Angew. Chem. Int. Ed., 2019, 58, 13149 (DOI: 10.1002/anie.201907488)
2. L Trump et al, Org. Process Res. Dev., 2020, 24, 734 (DOI: 10.1021/acs.oprd.9b00442)











No comments yet