A simple method for transferring point chirality to axial chirality
The idea of atropisomerism is not immediately obvious. This form of chirality arises when two forms of a molecule are differentiated by hindered rotation around a single bond, and gets its name from the Greek atropos (not turning). Chirality at a single atom (point chirality), in which two distinct geometric arrangements of four different groups around a central atom give non-superimposable structures, is definitely both a mouthful and conceptual challenge in its own right. But as a foundational tenet of chemistry, it is something students soon become familiar with. Atropisomerism is not only encountered less often, but is visually a more challenging concept: restricted rotation around a single bond is not something that can be definitively drawn on paper. We must mentally acknowledge that the bond in question cannot rotate. As a consequence, pairs of distinct, conformationally stable compounds are often viewed as one (figure 1).
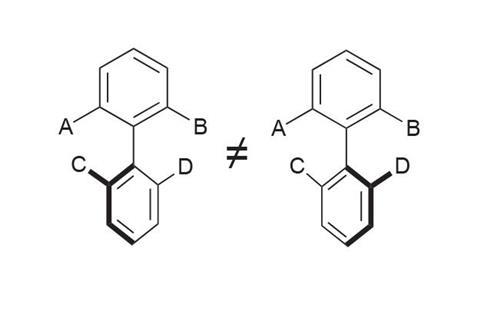
Atropisomerism is fequently observed in biaryls, giving rise to a plethora of chiral binaphthyl-derived ligands for a multitude of asymmetric transformations. But other structures – for example, tertiary amides and aryl groups within macrocycles – also display this behaviour.
In 2014, Christof Sparr and Achim Link of the University of Basel, Switzerland, demonstrated an innovative synthesis of atropisomeric binaphthyls using asymmetric enamine organocatalysis.1 An intramolecular aldol-based ring formation generates a chiral adduct that subsequently aromatises by eliminating water. The reaction proceeds with impressive transfer of the chiral information. Clearly inspired by this work, Damien Bonne, Jean Rodriguez and their team at Aix Marseille University, France, have developed a technique for preparing hetero-atropisomers containing 5-membered rings – an essentially unexplored field.2
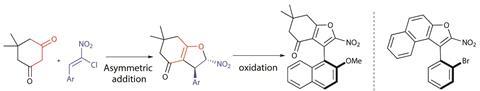
The team proposed that an asymmetric ring formation of substituted dihydrofurans would give access to molecules with defined chirality, which could direct a stereoselective oxidation, giving axially chiral furan derivatives (figure 2). Using a bifunctional squaramide organocatalyst, they began by adding dimedone (1,1-dimethyl-3,5-cyclohexanedione) to a substituted nitro-olefin. Conjugate addition and cyclisation gives the dihydrofuran on treatment with a little acid, in 92% yield and 95% enantioselectivity. It was then essential to find mild conditions to oxidise the partially saturated heterocycle and ensure efficient conversion of the point chirality in axial chirality. Using diacetoxyiodobenzene – a hypervalent iodine reagent – and CsCO3 in chloroform at –10°C gives the aromatic product with minimal erosion of the enantiomeric excess. This molecule proves to be remarkably stable with a rotational barrier of 122kJ/mol and a half-life of four years.
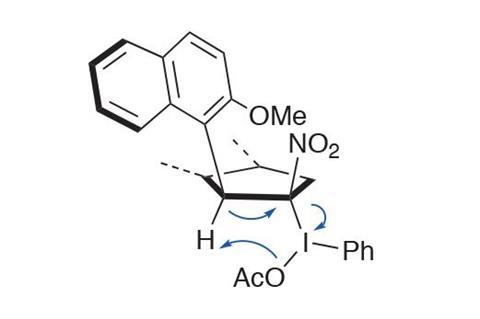
The researchers propose that the selectivity of the oxidation is controlled by the large methoxynaphthyl moiety: following deprotonation of the dihydrofuran to give the nitronate, the oxidant adds to the face opposite the naphthyl group. The subsequent elimination proceeds to minimise the interaction of the adjacent substituents by directing the larger aryl (vs methoxy) moiety away from the nitro group (figure 3).
Cyclic-1,3 diketones, -1,3 ketoesters and -1,3 ketoamides are suitable pro-nucleophiles, giving access to different furan derivatives. However, the team was likely surprised to find that, under the same reaction conditions, 2-naphthol also produces the corresponding dihydrofuran, as a product of a Friedel–Crafts like conjugate addition–cyclisation cascade reaction. With smaller aryl groups in place of the methoxynaphthyl group, oxidation with hypervalent iodine gives poor transfer of chirality. Switching to MnO2 in toluene at –5°C delivers 100% chirality transfer – but for the opposite axial enantiomer! This inversion of selectivity implies that the proposed model for stereocontrol is clearly not valid in this case. With 2-naphthol as the nucleophile, the team has explored a variety of aryl nitro-olefins as coupling partners, giving chiral products with rotational half-lives ranging from 12 days for methyl ester-substituted arenes, up to a million years with diphenylphosphine oxide.
References
1. A Link and C Sparr, Angew. Chem., Int. Ed., 2014, 53, 5458 (DOI: 10.1002/anie.201402441)
2. V S Raut et al, J. Am. Chem. Soc., 2017, 139, 2140 (DOI: 10.1021/jacs.6b11079)










No comments yet