





Explaining trends across the periodic table with the help of node-induced electron confinement
Chemical bonds are one of the first ideas we learn about in chemistry, where foundational concepts including electron sharing (covalency), transfer (ionicity) and delocalisation (metallicity) are used to understand chemical properties such as bond strengths, lattice energies, 3D structures, solubility and conductivity.
While on the surface these ideas seem well-established, an ongoing debate has been rumbling behind the scenes for over 100 years. With the discovery of the electron and the birth of quantum mechanics in the late 19th and early 20th century came questions about the wave-like nature of these quantum particles. Two camps formed, each with their own opinions on the origins of covalent bonding.
The first, initiated by John Slater,1 favoured the explanation that by increasing the density of electrons between positively charged nuclei, net electrostatic attraction holds atoms together. It is this explanation that is often first taught to chemistry students. The second camp formed around Hans Hellman’s quantum explanation,2 which hinges upon the wave-like behaviour of electrons: when an electron delocalises to be shared between two atoms in a chemical bond, its wavelength increases. Since kinetic energy depends inversely on wavelength, a longer wavelength of light corresponds to a lower energy photon, and the same is true of electrons. This energy lowering results in a more stable molecule, which we associate with bond formation.
To get to the heart of covalent bond formation, we need a way to understand how and why electrons become shared, and, crucially, what this sharing does to the energy of our molecule. Field-defining work from Klaus Ruedenberg beautifully demonstrated that electron delocalisation via kinetic energy lowering (an increase of wavelength) is responsible for covalent bonding in H2 and H2+.3 He also elucidated a second effect – orbital contraction – which describes a quantum mechanical mechanism by which the shared electron(s) in H2 and H2+ contract towards the nuclei to increase the strength of the bond. This contraction raises the (previously lowered) kinetic energy to obey the virial theorem, which connects changes in kinetic and potential energy to the length and strength of a bond.
Many years of dedicated work by scientists across the world have sought to understand whether this kinetic energy-lowering delocalisation effect, and in particular its relationship with orbital contraction, is universal to all covalent bonds. Unfortunately, no consensus was reached, with some studies suggesting that delocalisation and contraction were equally important for all covalent bonds,4 and others finding that contraction was only important for bonds involving hydrogen atoms.5
We recently devised a scheme to isolate the importance of each of these effects on bond formation, expanding from covalent bonds to also consider polar covalency and ionic bonding.6 By separating out the effects of delocalisation from changes in orbital contraction and polarisation (the change in shape of orbitals due to electric fields), we uncovered a previously overlooked effect that also controls chemical bond formation: node-induced electron confinement.
Our theory could predict whether a bond would be strengthened depending on the presence of valence orbitals with nodes
This phenomenon is not in itself new. We know that out-of-phase light waves interfere destructively, resulting in alternating regions of dark and light. The same phenomenon affects valence electrons as they attempt to delocalise to form a bond: when atomic orbitals with nodes overlap (for example, the 2s orbitals of a carbon atom), some parts interfere constructively, and some interfere destructively. This destructive interference decreases the wavelength of the electron, resulting in localisation (confinement) that increases its kinetic energy and weakens the bond. A second effect – Pauli repulsion – also thwarts the electron’s attempt to delocalise, in this case resulting from destructive interference between the electron involved in bonding and the inner electron shells of other atoms, since two electrons of the same spin cannot occupy the same region of space simultaneously.
Combining this pair of effects with orbital relaxation (contraction and polarisation) we could explain trends in bond strengths across the periodic table. Strong bonds form when delocalisation and orbital contraction can occur freely, such as in polar bonds where the removal of electron density from an electropositive atom allows significant contraction. Conversely, nodes in valence orbitals and the presence of core electrons result in weaker bonds.
We were particularly interested to understand what happens to bond strengths after an electron is removed from the molecule, and we were pleased to see that our theory could predict whether a bond would be strengthened or weakened depending on the presence or absence of valence orbitals with nodes. In addition, we found that molecules containing both s and p bonds, for instance N2, vary in strength according to the presence or absence of nodes in their p system.
With this unified theory of chemical bond formation that applies to bonds across the periodic table, we’re excited to further explore other types of important chemical interactions, for instance novel chemical bonds, as well as hydrogen, halogen and chalcogen bonds that find uses in catalysis, crystal engineering, supramolecular chemistry and beyond. We also hope that our work will clarify the importance of electron delocalisation during bond formation, and offer a teachable model for students first learning about the beauty and rich variety of chemistry that defines the world around us.
References
1 J C Slater, J. Phys. Chem., 1933, 1, 687 (DOI: 10.1063/1.1749227)
2 H Hellmann, Z. Phys., 1933, 85, 180 (DOI: 10.1007/BF01342053)
3 K Ruedenberg, Rev. Mod. Phys., 1962, 34, 326 (DOI: 10.1103/RevModPhys.34.326)
4 M Schmidt et al, J. Chem. Phys., 2014, 140, 204104 (DOI: 10.1063/1.4875735)
5 D S Levine and M Head-Gordon, Nat. Commun., 2020, 11, 4893 (DOI: 10.1038/s41467-020-18670-8)
6 A J Sterling et al, ChemRxiv, 2023, DOI: 10.26434/chemrxiv-2023-1lprg
Bonds are the ties that bind chemistry

Those seemingly simple sticks belie our most complex concept
- 1
- 2
- 3
- 4
- 5
 Currently
reading
Currently
reading
Towards a unified theory of bonding







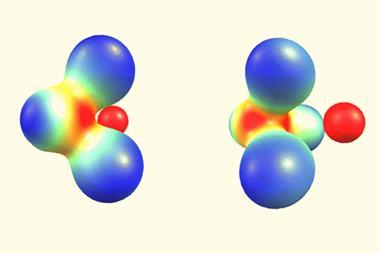









No comments yet