
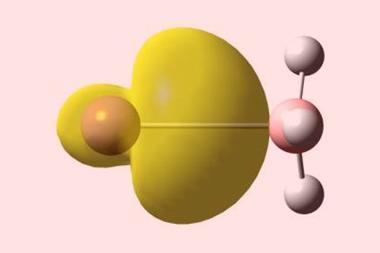
When chemists first synthesised NaBH3− back in 2019, initial research suggested a dative bond connected its sodium monoanion with a borane Lewis acid.1 Theoretical chemists, however, quickly established that the bonding was not that simple.
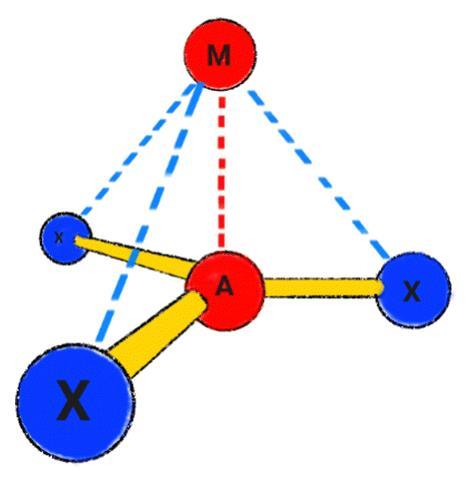
Since then, several computational studies have attempted to accurately describe the NaBH3− cluster. Sudip Pan, of Nanjing Tech University in China, and Gernot Frenking, of Philipps-University Marburg in Germany, reasoned that the metal–ligand bond was an electron-sharing covalent bond rather than a conventional dative bond.2 In contrast, Cina Foroutan-Nejad, from the Polish Academy of Sciences, suggested it was an ionic-enforced covalent bond driven by favourable interactions between the metal and the ligand’s substituents.3 Separate studies also proposed two-centre one-electron bonds,4 spin-polarised bonds5 and multifaceted metal–ligand bonds.6 Of these myriad bonding modes, ionic-enforced covalency, now recast as collective bonding, has emerged as a frontrunner.7
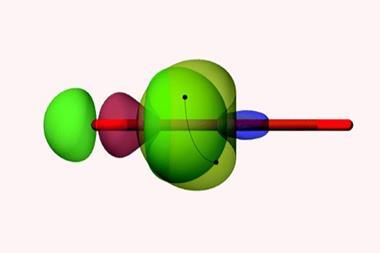
‘Classical bonds are attractive interactions between two neighbouring atoms,’ explains Foroutan-Nejad. ‘Collective bonds are different because the primary interaction between neighbouring atoms is repulsive.’ For organometallic molecules of the form MAX3, Foroutan-Nejad believes that stabilising M–X non-nearest neighbour interactions compensate for counterintuitively weakly stabilising, or even destabilising, nearest neighbour M–A interactions.
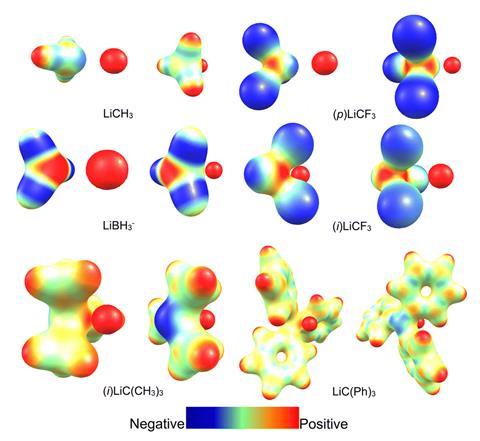
Now, a computational study led by Foroutan-Nejad and Ricardo Pino-Rios from Arturo Prat University in Chile, has employed electrostatic potential maps and the intrinsic bond strength index to corroborate their previous research on collective bonding.8 Electrostatic potential maps are pictorial tools that show the charge distribution within a system and help to identify attractive and repulsive interactions. The intrinsic bond strength index measures the chemical bond strength between a pair of atoms and accounts for quantum mechanical exchange–repulsion.9
From this study, Foroutan-Nejad and Pino-Rios established the circumstances under which collective bonds are present. They concluded that collective bonds predominate in MAX3 molecules with electropositive central atoms (A) and electronegative substituents (X), such as LiCF3. These molecules contain destabilising anti-electrostatic cation–π-hole and cation–lone-pair-hole (M–A) interactions, which are compensated for by the metal–substituent (M–X) interactions. In this context, a hole is a region of comparatively low electron density in a molecule.10
‘We tend to think of bonds as pairwise interactions however this is not always correct,’ comments Sebastian Kozuch, an expert in computational chemistry and chemical bonding from Ben-Gurion University of the Negev, Israel. ‘There are many examples in which one cannot pinpoint where the interaction comes from; these are very rare exceptions and this is why we have such huge discussions. My opinion is that these collective interactions are present.’
However, not all researchers agree. ‘There is always controversy about chemical bonding because it’s not well defined,’ affirms Miquel Solà i Puig, an expert in computational chemistry from the University of Girona in Spain. Solà had previously questioned whether collective interactions exist and remains unconvinced.11 ‘[This research] says that the lithium–carbon interaction is either destabilising or weakly stabilising and that all stabilisation comes from the substituents. I think that this is not true. Our [previous] results indicate that the covalent interaction between the lithium and the carbon is there and this is what common sense in chemical thinking would suggest. I’m not in favour of defining new types of bonding if it’s not totally justified.’
Whilst the debate over the bonding in these organometallic molecules seems set to continue, Foroutan-Nejad is hoping to implement his newfound understanding of collective interactions in real-world applications. ‘I am mainly thinking about catalysts,’ comments Foroutan-Nejad. ‘In a normal interaction the electrons of the central atom go toward the immediate neighbouring atom to form a bond. In collective interactions the electrons of the central atom are pushed away. This can be potentially used for designing new catalysts or perhaps understanding the mechanisms of action of some existing catalysts.’
References
1 G Liu et al, Angew. Chem., Int. Ed., 2019, 58, 13789 (DOI: 10.1002/anie.201907089)
2 S Pan and G Frenking, Angew. Chem., Int. Ed., 2020, 59, 8756 (DOI: 10.1002/anie.202000229)
3 C Foroutan-Nejad, Angew. Chem., Int. Ed., 2020, 59, 20900 (DOI: 10.1002/anie.202010024)
4 R Pino-Rios, D Inostroza, W Tiznado, Angew. Chem., Int. Ed., 2020, 60, 12747 (DOI: 10.1002/anie.202101403)
5 P Salvador et al, Angew. Chem., Int. Ed., 2020, 60 1498 (DOI: 10.1002/anie.202010948)
6 S Radenković, S S Shaik, B Braïda, Angew. Chem., Int. Ed., 2021, 60, 12723 (DOI: 10.1002/anie.202100616)
7 S Sowlati-Hashjin et al, Nat. Commun., 2022, 13, 2069 (DOI: 10.1038/s41467-022-29504-0)
8 R Pino-Rios et al, Chem. Commun., 2024, 60, 400 (DOI: 10.1039/d3cc05451a)
9 J Klein et al, J. Phys. Chem. A, 2020, 124, 1850 (DOI: 10.1021/acs.jpca.9b09845)
10 R Shukla et al, Phys. Chem. Chem. Phys., 2023, 25, 12641 (DOI: 10.1039/d3cp00225j)
11 J Poater et al, Nat. Commun., 2023, 14, 3872 (DOI: 10.1038/s41467-023-39498-y)












No comments yet