
As the clouds clear on computational crystal structure prediction, is the technique ready to empower mainstream materials research? James Mitchell Crow reports
-
Importance and challenges: CSP is crucial for understanding and developing materials with specific properties, such as pharmaceuticals and photovoltaics. The process involves predicting how molecules pack together in crystals, which is complex due to the vast number of possible structures and the need for highly accurate energy calculations.
-
Historical progress: Initially, CSP relied on force fields to estimate interactions between atoms, but this approach struggled with accuracy. The breakthrough came with the use of density functional theory (DFT), which provided more precise predictions but at a high computational cost.
-
Applications and successes: CSP has been successfully applied in the pharmaceutical industry to predict polymorphs and avoid issues with drug stability. It has also been used to design new materials with specific properties, such as porous materials for gas storage and photomechanical crystals that change shape under light.
-
Future directions: To make CSP more accessible and efficient, researchers are exploring the use of artificial intelligence (AI) and machine learning to reduce computational costs. New methods, such as the CrystalMath protocol, aim to simplify the prediction process by identifying topological rules from existing crystal structure data.
This summary was generated by AI and checked by a human editor
With the firing of a virtual starting gun, theoretical chemists around the world cracked their knuckles, filled their coffee pots, and fired up their high-performance computers.
Participants in the latest Blind Test of Crystal Structure Prediction (CSP) were given a handful of 2D organic molecular structures – and exactly one year. The task was to predict each molecule’s likely crystal structures, juggling virtual atoms into position to simulate the orderly packed structure that the molecules adopt as they crystalise.
At the seventh blind test’s close, efforts would be judged against the target molecules’ experimentally solved crystal structures – an unpublished secret until the test’s end – determined from crystals grown in the lab.
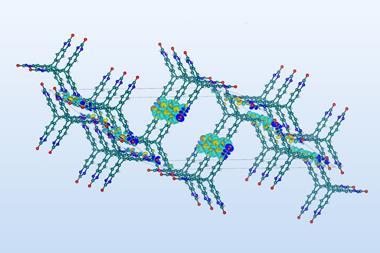
The large number of participants from industry underscored CSP’s real-world importance. From pharmaceuticals to photovoltaics, many functional organic materials are based on molecular crystals. Many of their properties, from stability and bioavailability to colour and conductivity, stem at least in part from the way their molecules pack together when they crystallise.
The latest blind test was the most challenging yet. ‘What stuck with me was a comment from an experimentalist, saying “This is the first test where the target molecules look like things that I work with,”’ says Gregory Beran, a theoretical chemist working on CSP at the University of California Riverside, US. By the end of the test, would the participants’ crystal structure predictions also look like the real thing?
Use the force fields
In the lab, coaxing organic molecules to crystalise can be slow and difficult. Adding to the challenge, many organic molecules are ‘polymorphic’, able to pack into multiple distinct crystalline forms. Even seemingly cooperative compounds can hide undiscovered polymorphs, whose sudden appearance could derail the launch of a much-needed new pharmaceutical, for example.
CSP would be a powerful tool to guide these efforts, generating virtual maps to aid molecular crystal landscape exploration. The maps could help to place experimental crystal structure and property findings into context – and even signpost areas for new materials discovery.
For decades, however, calculating crystal structures seemed an intractable problem. In 1988, the frustration was expressed by Nature’s then-editor John Maddox. ‘One of the continuing scandals in the physical sciences is that it remains in general impossible to predict the structure of even the simplest crystalline solids from a knowledge of their chemical composition,’ he wrote.
People typically go about the problem by looking for local energy minima on an energy surface
‘Crystal structure prediction is one of those things that seems like it should be simple, until you really think about all the challenges that it poses,’ Beran says. For any given organic molecule, the process typically involves first randomly generating a broad set of possible crystal packing structures. ‘The first challenge is that this search space is huge,’ Beran says. A random structure generation step can produce 105 to 107 structures.
To predict which of these myriad structures might ever appear in an experimentalist’s flask, the next step is to calculate each structure’s relative energy and identify the most stable molecular packing arrangements. ‘People typically go about the problem by looking for local energy minima on an energy surface based on the degrees of freedom that define a crystal structure,’ says Graeme Day, a CSP specialist at the University of Southampton, UK.
With even the simplest crystal structure possessing 12 degrees of freedom, however, these energy surfaces are mind-bendingly high dimensional. ‘Finding the local minima on a 12-dimensional energy surface is a big problem, and most things we’re looking at are even higher dimensional than that,’ Day says. Additional degrees of freedom arise in molecules with flexibility such as rotatable bonds, or with more than one molecule in the crystal.
The next layer of challenge is that the computational modelling must be extremely accurate to correctly rank the energetics of each structure. ‘Once you get these local minima, you want to figure out which is the deepest – which has the lowest free energy,’ says Day. ‘And they turn out to be very closely spaced, sometimes separated by fractions of a kilojoule per mole.’ The vast number of potential structures to be evaluated compounds the difficulty of accurately ranking them.
You have all these interactions in structures that end up being almost at the same energy
Since it was first run in 1999, the CSP blind test has offered the community a benchmark of its progress against this nested set of problems. During the first few blind tests, the method of choice was to deploy force fields, collections of equations that predict the most stable crystal structure by estimating forces between atoms.
Accurately ranking the energetics of crystal structures separated by very fine margins, when there are numerous finely balanced forces at play, is a formidable computational challenge. ‘There’s a subtle competition and interplay between factors including hydrogen bonding, dipole–dipole interactions, pi stacking – which are also often in competition with intramolecular conformation,’ Beran says. ‘You have all these interactions, some additive and some cancelling out, in structures that end up being almost at the same energy.’
Even though the first few blind tests set only small, simple, rigid molecules as targets, the number of successful predictions was soberingly small. ‘Force fields really struggled with regards to the intra- versus inter-molecular forces, and just the number of different types of interactions involved,’ Beran says.
A higher power
When Day began his independent research career in the mid-2000s – almost two decades after Maddox had decried the lack of progress – some were questioning theoreticians’ entire line of attack. ‘The quite low hit rates in the first few blind tests was giving people outside of the field the impression that we weren’t getting anywhere,’ says Day. ‘People would tell me “You must be taking the wrong approach – maybe it’s not about ranking energies, maybe it’s all about the kinetics of crystal growth.”’
The breakthrough came in 2007, when a team in the Fourth Blind Test that included Marcus Neumann from Germany-based CSP software specialist Avant-garde Materials Simulation added an extra layer of modelling firepower. After using force fields to roughly rank the candidate structures, the team reanalysed the top few candidates using density functional theory (DFT), a much more computationally demanding method based on quantum mechanical modelling. Deploying DFT, they got every prediction correct.
The finding confirmed that ranking energies had been the right approach all along, says Day. ‘People saw that if you can afford to apply very accurate methods, then this approach works,’ he says. Even so, the first wave of DFT-predicted molecular crystal structures were based on small and simple molecules, far from the real-world cases keeping experimentalists up at night.
It’s taken another decade or two, but we’re reaching the point where, even for complex drug molecules, successes are expected
‘The first crystal engineering conferences I went to, around 2010, there was a lot of scepticism from people on the experimental side about whether we were ever going to be useful,’ Beran says. ‘I think that has now changed dramatically.’
Refinements to DFT-based CSP methodology have continued – such as the correction developed by Beran to account for DFT’s tendency to over-estimate electron delocalisation in many organic molecules. ‘It’s taken another decade or two, but we’re reaching the point where, even for complex drug molecules, successes are expected, and useful information is gained more often than not,’ Beran says.
One sector to embrace and drive CSP advances has been the pharmaceutical industry, to assist in new drug solid form screening. In the past, the sudden appearance of an unexpected drug molecule polymorph – which can change key properties such as solubility and stability – has derailed the launch or triggered the withdrawal of medicines for conditions from HIV to Parkinson’s. CSP is now widely used to help warn of possible pharmaceutical polymorphs not detected during experimental screening.
Other sectors are beginning to embrace the technology too. ‘In the early days of DFT, we’d apply it to systems where the crystal structures were already known, to see if we could “predict” them – which is useful for method development, but not for convincing the wider community,’ Day says. ‘It takes those cases where you decide what molecules to make based on predictions, and find a new structure that had been predicted.’
There has been a growing number of those moments, Day adds. ‘There have been some pharmaceutical cases, and cases with functional materials – such as the porous materials research that I’ve been involved in.’
Unblocking pores
Working with experimentalist Andrew Cooper at the University of Liverpool, UK, Day and his team predicted the structure of a new set of porous crystals, and also predicted their capacity to store methane or to selectively separate mixtures of hydrocarbons within their pores.

Usually, stable organic crystals are those in which the molecules achieve the densest possible packing. The team showed that, by crystalising their predicted porous molecule with guest solvent molecules within their pores to stabilise the structure, they could make several of their proposed structures in the lab. These organic crystals’ experimentally determined properties proved to be a good match for their predictions.
Beran is part of a team working to predict and make crystals with powerful photomechanical properties. ‘The molecules in these organic crystals are packed in a way that they can undergo a photochemical reaction in the solid state,’ Beran says. When exposed to light, the photochemical reaction results a physical change to the crystal – such as bending, jumping, elongation or compression – with the potential to do useful work.
‘There are examples of milligrams of photomechanical materials lifting hundreds of grams of weight,’ Beran says. ‘We’ve found that changes in the crystal packing of a given molecule can have a huge impact on the photomechanical response,’ he adds.
Polymorphs in which molecules pack in parallel, so that each molecule’s photochemistry-driven shape change is aligned along a single axis, generates the largest displacement in the overall crystal. The team has discovered examples where the same molecule can generate a 40-fold higher work density by packing as one polymorph rather than another. ‘It’s a difference at least as large, if not larger, than the classical crystal engineering approach of looking at different photochemical reactions or different molecules,’ Beran says.
Such studies tease the kind of contribution that CSP could make for new materials discovery. The blocker is that DFT-based CSP comes with very high costs that, if anything, made the technique increasingly less accessibly, not more, Beran says.
We’re pushing the complexity of systems we can apply crystal structure prediction to
In the latest blind test, the most successful teams came from companies that employed dedicated specialists from structure search through to quantum chemistry and free energy simulations. ‘As we get to these complex systems, it’s harder and harder for a single domain expert to do everything,’ Beran says. Even for academic specialists, cutting edge CSP is becoming more difficult.
The other challenge is the computational resources that DFT demands, which few outside of the pharmaceutical sector can afford. Compared to force fields, DFT energy calculations can be 103 to 105 times more computationally expensive, says Day. The associated supercomputer carbon emissions are not trivial, and the slow nature of the calculations severely limits the number of CSPs that can be performed. ‘The eight or nine molecules in our initial porous materials study took a few months of our allocation on Archer, the UK’s national high-performance computer,’ says Day.
The time it takes to run the numbers curtails the utility of the technique, Day adds. ‘We’re pushing the complexity of systems we can apply CSP to, to a point where the predictions are sometimes slower than just doing the experiments.’
Cheap tricks
To break apart the bottleneck of DFT reliance, one option – now being explored by Day and his team – could be to make CSP faster and more useful with AI. ‘We’ve been training models to predict the energetics of crystals to nearly DFT level accuracy, but at fractions of the computational cost,’ says Day. ‘Just last year, using machine learning, we published work where we did 1000 molecules, and we are now working on stuff which is multiple thousands of molecules.’
This level of acceleration broadens the range of molecules that can be explored in the hunt for new organic materials with target properties, Day says. ‘In our DFT-based porous materials work, because we could only look at around 10 molecules, we had to have a hypothesis about the kind of molecules we wanted to target,’ he says. ‘Now, with the help of machine learning, we can be a lot more open with the chemical space that we explore.’
The best machine learning method to apply in different CSP scenarios is one question that Day is investigating. Machine learning could directly learn how to model the relationship between the geometry and the free energy of a crystal structure. Alternatively, a quicker and more accurate approach may be to pair machine learning with force fields. Known as delta machine learning, this uses models trained on the difference between the relative energy rankings generated by force fields and the rankings generated by higher level theory such as DFT. The trained model can then quickly correct the output of an inexpensive force field model into a ranking approaching DFT accuracy.
There’s even the possibility to conduct iterative delta learning, where a second model compares the output of the first model against actual DFT results, and learns how to apply a second round of correction. Each iterative step is an easy lesson for machine learning to master. ‘Delta learning is going to be useful because we will need less training data to get that correction to a reference energy,’ says Day.
AI has great promise, says Beran, but it’s still early days. ‘I feel like there haven’t yet been clear home runs where machine learning has shown a transformative impact – but I think it’s coming,’ he says. The latest hot topic in AI, generative models, are also being explored, including to propose new molecules to analyse by CSP (see box Inorganic generation below].
To cut CSP’s computational cost, AI isn’t the only strategy being pursued. The vast stores of experimentally derived crystal structure data, captured in resources such as the Cambridge Structural Database, can also be interrogated by human brains in search of previously unrecognised rules, according to work by Mark Tuckerman from New York University in the US.
In late 2024, Tuckerman published a computational protocol called CrystalMath, which predicts crystal structures by applying a set of simple topological rules divined from the CSD. When the team applied the method to one of the more challenging structures from the sixth blind test – for which DFT had consumed millions of CPU hours to identify only some of the molecule’s known polymorphs – it took CrystalMath 32 hours to predict all five known polymorphs, running on a mid-range laptop.
Inorganic generation
Inorganic crystals consist of assemblages of atoms or ions, rather than of molecules, posing its own set of challenges for inorganic crystal structure prediction (CSP).
‘In organic crystals, you’re looking at quite weak interactions between molecules that are very difficult to quantify – but you know what the molecule itself looks like,’ says Volker Deringer, a computational inorganic chemist at the University of Oxford, UK. ‘Whereas in inorganic solids, you sometimes don’t even know the local coordination environment.’
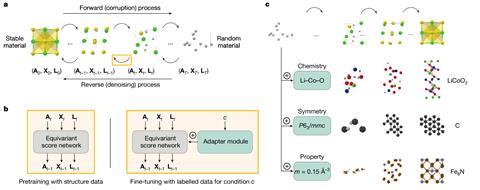
Differences aside, inorganic and organic CSP has involved the same basic process of generating putative structures for a given compound and then using DFT to rank their energies. ‘DFT-based CSP has been extremely successful for elemental systems, with alkali metals being prominent examples, and binary phases have also been widely explored,’ Deringer says. But DFT quickly becomes computationally unaffordable for more complex chemical compositions, he adds. ‘Once you go to four-component systems, which is common in materials chemistry, it gets very complicated’ he says.
In the inorganic sphere, efforts to overcome this limitation by deploying machine learning as a faster, leaner alternative to DFT are well underway. ‘Inorganic chemists are a bit ahead with replacing or speeding up the DFT step with machine learning for CSP,’ says Graeme Day, a CSP specialist at the University of Southampton, UK. ‘Inorganic crystals have these families of common structures, so the machine learning model is going to see similar structures over and over in training, which might help.’
Inorganic chemists and AI specialists are also breaking ground in applying generative models for predicting inorganic crystals with desirable target properties. In January 2025, Microsoft published a generative model called MatterGen, which reportedly generates putative inorganic materials that are twice as likely to be novel and stable than previous generative models, and can propose novel materials with desired mechanical, electronic and magnetic properties.
‘Generative models are about likelihoods: based on lots of structures I’ve seen, what would be another likely one?’ Deringer says. ‘These generative models for crystal structures are one of the current frontier methodologies, much less widely used so far than direct simulations with DFT or machine-learned potentials, but they can predict interesting things and are quite exciting for predicting the next material to try to make in the lab.’
Ground rules
Tuckerman’s work was spurred by the agonisingly time-consuming nature of conventional quantum mechanical modelling approaches, he says. ‘Sometimes it works, sometimes it doesn’t – and meanwhile, your experimental colleagues always seem to be two years ahead of you,’ he says.
The new approach was inspired by a 1967 paper written by mathematician and crystallographer Jakob Burckhardt on the discovery of the 230 space groups, which describe the rotational and translational symmetry of the molecular packing within a crystal. ‘He put forward the idea that maybe crystal structures could be predicted purely mathematically,’ Tuckerman says. ‘There’s this is beautiful theory of space groups, so maybe there are other mathematical rules that govern how molecules pack into crystals – so we downloaded the Cambridge database and started asking a lot of questions of it.’
The process took five years, Tuckerman says. ‘But when you start to ask the right questions, you see that there are patterns, which obey fairly simple rules – from which we could come up with a scheme to predict crystal structures on your laptop.’
One rule relates to the orientation of axes that predict where most of the molecule’s mass will be located. ‘Crystals with the highest density, where the molecules are most efficiently packed, tend to be the most favoured – which doesn’t leave a lot of room for molecules to move around,’ Tuckerman says. ‘When you think “OK, molecules in stable crystals don’t have much rotational freedom,” already you can discover one of the rules that’s really well obeyed by the database.’
We think we can do really high quality prediction in just tens of thousands of CPU hours, instead of million
In stable structures, the team found, the three principal axes of rotation will be oriented orthogonal to a commonly used crystal structure descriptor called crystallographic planes. In well-packed stable crystals with minimal rotational freedom, these axes should be good predictors of where the molecule’s mass is, Tuckerman says. ‘It makes sense that you couldn’t rotate things around these axes because there’s too many other atoms to bump into.’
‘I think the work on topological crystal structure prediction is amazing,’ says Beran, who recently submitted a research proposal with Tuckerman to further explore the idea.
As well as being fast, the technique is selective, predicting only small numbers of polymorphs – potentially overcoming force fields’ and DFT’s problem of ‘over-predicting’ numerous putative structures that are never observed experimentally. ‘I think the reason is that our rules are inspired by known crystal structures – and so, if the rules apply, you should get experimentally realisable structures,’ Tuckerman says.
Beran and Tuckerman plan to explore the idea of combining CrystalMath predictions with high level computation to complete the energy ranking step. Because of the small number of putative structures generated, the team could go beyond DFT to apply even higher levels of theory. ‘We think we can do really high quality CSP in just tens of thousands of CPU hours, instead of millions,’ Beran says.
There’s also the possibility that putative structures could be ranked without deferring to high level quantum theory at all, simply by assessing how well each structure obeys the topological rules, Tuckerman says. ‘I think there are more rules to be discovered,’ he adds. ‘For example, we found out very recently – again by examining the database – that intramolecular bonds between heavy atoms tend to lie within certain predictable planes.’ The team is also exploring the technique’s ability to predict the structure of challenging cases such as very flexible molecules, co-crystals, salts and hydrates.
‘If CrystalMath holds up to its preliminary promise, I think there’s a lot of potential to transform CSP and take it beyond something only companies with pharma-sized budgets can do,’ Beran says. ‘CSP is too niche at the moment – there is a very small number of people in the world who can do it well,’ he adds. ‘If we can make it accessible to everyone, the impact of this field could be much, much larger than today.’
James Mitchell Crow is a science writer based in Melbourne, Australia
Article updated 24 April 2025 to correct the name of the Cambridge Structural Database













No comments yet