
The 2013 Nobel prize in chemistry was awarded to three computational pioneers who combined quantum and classical mechanics. Emma Stoye learns about the latest laureates
For many of today’s scientists, computers and the programs they run are as vital to modern research as any lab equipment or reagent. Chemists are no exception, and over the last 50 years computers have probed the structures of molecules and proteins, predicted the outcomes of chemical reactions and identified targets for new drugs.
In recognition of this, the 2013 Nobel prize in chemistry was awarded to Martin Karplus of Harvard University, Michael Levitt of Stanford University and Arieh Warshel of the University of Southern California for, as the committee put it, ‘taking the experiment to cyberspace’.
Beginning in the 1970s, the three US-based laureates developed molecular modelling techniques that decades later still underpin simulation software used by scientists around the world. ‘Warshel, Levitt and Karplus completely revolutionised the field and changed the way we think about biological systems,’ says Lynn Kamerlin, a theoretical chemist at the University of Uppsala in Sweden, who was a postdoctoral researcher in Warshel’s group. ‘At a time when people were just starting to use computers, they were brave enough to go after the kinds of discoveries that seemed more like science fiction.’ Laying the foundations of modern molecular modelling was a team effort that required passion, persistence and ambition.

Enzymes on punch cards
‘Computers can actually solve anything if you just know how to put the question to them,’ Warshel tells Chemistry World. But this has often proved easier said than done, particularly in the late 1960s, when nothing resembling today’s home computers existed. The room-sized ‘supercomputers’ found at the world’s best institutions achieved a mere fraction of the speed, processing power and memory of modern smartphones. Warshel remembers programming as a completely different experience.
‘You had to carry this box of punch cards everywhere – and there was a risk that they would fall down and get mixed up,’ he says. ‘Often to get any results you had to come back to the computer at midnight because the turnaround was so slow!’
To our surprise, we were actually able to do something with whole proteins
Michael Levitt
Warshel first began to work with computers while studying chemical physics under the late Shneior Lifson at the Weizmann Institute of Science in Israel – a career choice he describes as ‘almost accidental’. Lifson’s lab was exploring new ways to figure out the structures of different molecules, with the help of the institute’s Golem supercomputer (which had less than 1MB of memory). Early computer models were treated in a similar way to the physical models structural scientists used to make from wire and plastic. The calculations that built them were based on classical mechanics – rules based on Newton’s laws of motion that treated atoms and the bonds between them as if they were balls connected by springs. Different calculations could be used to explore the energy and strain of the ‘springs’, and to consider electrostatic effects and van der Waals forces.
During his PhD studies at the Weizmann, Warshel met Levitt, who was there as a summer student before starting a PhD in the UK. Having learned to write computer code at classes and summer schools, Levitt helped Warshel make programs that modelled the basic properties of molecules.
‘Initially we were working with small molecules – about 50–70 atoms,’ Levitt tells Chemistry World, ‘but then I realised one program could be modified to deal with much larger ones. To our surprise, we were actually able to do something with whole proteins.’ By modelling two proteins whose structure had recently been solved – myoglobin and lysozyme1 – the group laid the groundwork for more complex simulations.
A marriage of quantum and classical
While these ‘molecular mechanics’ calculations could deal with large structures, they could only model their resting states, which made it impossible to study the conformational changes or dynamic behaviour of proteins. Calculations that describe the breaking and making of chemical bonds need to consider not just atoms, but the electrons and nuclei they are made of. This had traditionally been the domain of the physics community, who had made programs based on quantum mechanics. But the calculations took an enormous amount of time and processing power, so they were limited to a few atoms. Dealing with whole proteins was unthinkable.
‘At that time, each kind of model had specific limitations,’ explains Chris Cramer, a computational chemist at the University of Minnesota in the US. ‘The molecular mechanics ones which dealt with larger molecules were useful for simple functions like molecular geometries and energies of rotations to new conformations, but lacked any information about electrons. Quantum mechanics, on the other hand, was wonderful for bonding and electronic spectroscopy, but was so expensive that it couldn’t really be applied to much of anything.’ The real turning point, he adds, was bringing the two together.

In 1970, Warshel became a postdoc in Karplus’ group at Harvard. Karplus had spent many years developing programs based on quantum mechanics, while Warshel had experience of classical calculations. Together they created a program that could calculate the energy changes of electrons within the p-bonds of simple planar molecules such as 1,6-diphenyl-1,3,5-hexatriene. The program used quantum mechanics to model the effects of p-electrons, and classical mechanics on the atomic nuclei and s-electrons.2 Thus, they demonstrated for the first time that it was possible to combine quantum and classical mechanics in a single model.
Warshel continued to develop these hybrid approaches throughout the 1970s, travelling to the UK Medical Research Council’s Laboratory of Molecular Biology in Cambridge, where he was reunited with Levitt. The institute was already home to several Nobel laureates. ‘Those years were amazing,’ remembers Levitt. ‘I was so lucky to be able to work there alongside people like Francis Crick and John Kendrew – these heroes of science.’
The pair were ambitious, setting their sights on modelling a more complex system than ever before: an enzyme reaction. It was clear that a hybrid approach would be needed – the reaction involved making and breaking bonds, but the reactive site was part of a large protein containing thousands of atoms.
In 1976, they published their simulation of the lysozyme enzyme cleaving a glycoside chain.3 Their model treated atoms within the enzyme’s active site with quantum mechanics, but dealt with the rest of the system more efficiently using molecular mechanics. It wasn’t easy: ‘As well as worrying about the enzyme we had to worry about the solvent,’ says Levitt. ‘And of course we had to get all these different calculations to recognise each other and work well together.’
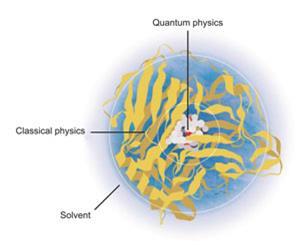
The solution was to incorporate energetic coupling terms to model the interaction between the classical and the quantum parts of the system, and get these parts to interface with the surrounding solvent. Distant parts could even be simplified further by essentially lumping together the atoms of individual amino acid residues and treating them as single units.4 This approach – now referred to as quantum mechanics/molecular mechanics (QM/MM) – has allowed ever bigger and more complex chemical systems to be simulated as computers have got faster and cheaper.
Researchers now model ion channels, viruses and antibodies using QM/MM. And its applications aren’t limited to biological systems. ‘One of the things my group is interested in is energy transfer from dyes into nanoparticles,’ says Cramer. ‘This is relevant to solar energy capture. You need to excite a molecule, which injects an electron into the semiconductor it’s attached to. You have to have quantum mechanics for that, but nanoparticles are big, so it’s helpful to have at least part of them be represented by molecular mechanics.’ In the 1970s, about 100 computational chemistry papers were published each year. Today, that figure is closer to 100,000.
Bringing experiment and theory together
This year’s Nobel prize honours more than any one breakthrough or technique. The laureates’ work kickstarted an important cultural shift in chemistry research, where theoretical studies came to be valued just as much as experiments.
‘The bringing together of experimental data and theoretical computational analysis has completely transformed a lot of structural biology,’ says Chris Dobson, a chemist at the University of Cambridge in the UK, who first collaborated with Karplus while on sabbatical at Harvard in the late 1970s. ‘But at the time they started doing it, it was thought to be a bit wacky, to put it mildly.’
Nowadays, thousands of people are using calculations based on the methods we began to develop in the 1970s
Martin Karplus
Dobson’s group is currently exploring protein folding and misfolding using both experiments and molecular models. ‘Simulations can be used to define reaction intermediates that can’t be defined experimentally because they’re too short-lived,’ he says, ‘and incorporating experimental data directly into simulations as restraints can have some really dramatic effects.’
Karplus also feels there was early opposition to the field. ‘When we first began doing [this] type of work, my chemistry colleagues thought it was a waste of time, [and] my biology friends said that even if we could do this, it wouldn’t be of any interest,’ he told a news conference after the prize announcement. ‘Nowadays, thousands of people are using calculations based on the methods we began to develop in the 1970s. I think that the Nobel prize, in a way, consecrates the field.’
Business models
One of the most significant impacts of computational models has been in the pharmaceutical industry, where they form an important part of drug development. ‘Models are used in the “lead optimisation” phase of drug discovery, when small changes are made to a molecule in order to improve its properties, such as affinity for the protein we are targeting, solubility and safety,’ says Darren Green, director of computational chemistry at GlaxoSmithKline. Predicting the effects of these changes using models shortens the whole process, cutting the time it takes to get new drug candidates ready for clinical trials.
‘Models are [also] used for “virtual screening” to identify promising compounds,’ says Green, ‘and they assist our development work, for example, by predicting the properties of the crystalline drug form that will end up in a pill.’
Many modern software packages still rely on the basic principles established by this year’s laureates, according to Adrian Stevens from Accelrys, a company that develops modelling tools for pharmaceutical companies. Some tools, for example, are based on CHARMM (chemistry at Harvard macromolecular mechanics) – a molecular dynamics simulation and analysis program that was first developed by Karplus’ group decades ago. CHARMM calculates force fields – mathematical functions used to describe the energies of atoms and molecules within different parameters – and can use these to simulate the structures and behaviour of molecules.
‘At Accelrys, CHARMM has underpinned our molecular modelling tools for around 30 years,’ says Stevens. ‘It still provides the framework for us to do simulations.’
Chemistry’s next top model
For a field that has progressed so much over the last few decades it is difficult to predict what the future holds. Karplus, Levitt and Warshel still oversee research programmes at their institutions. Warshel says he is now in ‘the most productive time of my career’.
‘While I’m really excited about the Nobel prize, I’m also really excited about all the work we’re doing right now,’ says Levitt, whose group applies computational methods to diverse problems in biology. ‘And I’m hoping that now I can carry on helping young scientists even more than I have before.’
Between them, all three laureates have trained scores of students and postdocs who now populate labs around the world. Kamerlin says Warshel was a great mentor to her – and still is. ‘He’s a brilliant scientist,’ she says, ‘but he also has a wicked sense of humour, and still has time to watch mindless TV with his grad students.’
Looking ahead, she says computational chemists will not only model bigger systems, but will be able to do so using fewer simplifications. This is something her own group is working on. ‘Now we have the computing power to really understand what’s going on,’ she says. ‘So instead of bigger and bigger, we’re trying to go deeper and deeper.’
In 2001, Levitt hypothesised that one day we will be able to simulate an entire organism using its genomic sequence.5 But is he still as optimistic 12 years later? ‘That statement may well have come from a little bit of euphoria left over from the millennium!’ he says. ‘But ultimately, this is computers. So you never know.’
References
1 M Levitt and S Lifson, J. Mol. Biol., 1969, 46, 269 (DOI: 10.1016/0022-2836(69)90421-5)
2 A Warshel and M Karplus, J. Am. Chem. Soc., 1972, 94, 5612 (DOI: 10.1021/ja00771a014)
3 A Warshel and M Levitt, J. Mol. Biol., 1976, 103, 227 (DOI: 10.1016/0022-2836(76)90311-9)
4 M Levitt and A Warshel, Nature, 1975, 253, 694 (DOI: 10.1038/253694a0)
5 M Levitt, Nat. Struct. Biol., 2001, 8, 392 (DOI: 10.1038/87545)












No comments yet