
Andy Extance looks into the latest in Alzheimer’s disease, pain and memory
Our brains are massively complicated – and so too are the biggest biochemical issues affecting them. Here we look to simplify the big open questions in neurochemistry and fuel your curiosity about three critical areas: Alzheimer’s disease, arguably the most feared ageing-related disorder; pain, one of the poorest-understood biochemical processes involving the nervous system; and memory, including the recent discovery of a new chemical process involved in storing information. This explainer will look at the current understanding, the unanswered questions, the techniques and chemistry involved, as well as future prospects.
Alzheimer’s disease
How does it work?
The amyloid theory of Alzheimer’s disease originates from Alois Alzheimer’s 1906 post-mortem analysis of a woman’s brain and the hallmark plaques and tangles it contained.
In the decades since, scientists have determined that the plaques consist of a small protein called beta-amyloid, produced when amyloid precursor protein (APP) is cleaved by protease enzymes. Excess beta-amyloid collects in the brain and eventually aggregates. The tangles consisted of a second protein called tau, which usually helps give neurons their shape.
The lack of a genetic link between tau and Alzheimer’s disease has been a conundrum
In the early 1990s, researchers found mutations in the gene for APP that led to people developing Alzheimer’s disease relatively young, explains Karen Duff, director of the UK Dementia Research Institute centre at University College London. That led people to focus on beta-amyloid as the primary cause of illness in the disease.
Around the same time German anatomists Heiko and Eva Braak looked at the number and distribution of amyloid plaques and tau tangles in thousands of people who died at various stages of Alzheimer’s disease. ‘Tangle pathology correlated with when they first showed cognitive impairment, but it wasn’t just the number of the tangles, it was also where they were distributed,’ says Duff. ‘The link with tau tangles was much stronger than the link with amyloid plaques, the number of them and where they were distributed.’
Positron emission tomography (PET) studies that image the brains of Alzheimer’s’ patients have confirmed the Braaks’ observations, Duff says. ‘The distribution of tau correlates well with initiation of cognitive impairment,’ she says. Yet in contrast to APP, no mutations that cause Alzheimer’s disease have been found in the tau gene, Duff stresses. ‘The lack of a genetic link between tau and Alzheimer’s disease has been a conundrum,’ she says.
The prevailing hypothesis is therefore that beta-amyloid triggers the start of the disease, Duff explains. ‘Amyloid has now been shown to accumulate for quite a long time before any cognitive impairment is seen, something like 10 to 15 years,’ she says. Cognitive impairment, however, is caused by abnormal forms of tau accumulating, which ‘is clearly related to cell death’, says Duff, ‘whereas accumulation of amyloid is not clearly related to cell death’.
What have we learned in recent years?
The US Food and Drug Administration (FDA) approved the beta-amyloid targeting drug aducanumab in 2021. Developing drugs that reduce amyloid plaques in the brain was a ‘scientific breakthrough with major implications’, according to Paresh Malhotra from Imperial College London, UK. It showed that drugs based on antibodies like those our immune systems use to remove potentially disease-causing invaders from our bodies can also remove beta-amyloid. Malhotra, whose team uses PET scanning for beta-amyloid extensively in National Health Service patients, highlights a 2016 PET study showing how aducanumab reduces beta-amyloid levels in the brain.
Malhotra also highlights two clinical trials showing that ‘amyloid clearance’ appears to translate to slowing Alzheimer’s disease progress. The trials were for two antibody drugs, lecanemab, approved in 2023, and donanemab, which is awaiting a decision from the FDA. ‘There’s controversy about how meaningful the clinical response is,’ Malhotra says. ‘But nonetheless, the changes in clinical status are in the correct direction. There is a reduced decline in people being treated with the amyloid-based drugs across multiple measures. And so, I think that represents a major breakthrough.’
The approval ‘was a great day for science because it showed us we were on the right track’, says Duff. ‘But in my opinion, not such a great day for patients, because the effect in humans was small.’ However, she is confident that with further improvements in the antibody approach and developing other drug strategies ‘results for patients will improve and it will become a more justifiable approach’. Meanwhile, targeting tau proteins with antibodies has been unsuccessful so far, Duff notes.
What role is chemistry playing?
Over the last 20 years, researchers have developed small radioactive ligand molecules that accumulate either in amyloid plaques or tau protein to make them visible in PET scans for Alzheimer’s. The first such ligand was developed in 2004, a carbon-11-containing radioactive analogue of a fluorescent dye, known as Pittsburgh compound B (PiB). However, its half-life was too short for clinicians like Malhotra to use, he explains. As a result, researchers have developed three fluorine-18 containing ligands, florbetapir, florbetaben and flutemetamol, which are better suited to detecting beta-amyloid in medical settings. ‘Amyloid PET is just about gold standard for demonstration of amyloid in the living brain,’ says Malhotra.
What does the future hold?
Because there is much evidence that people can have amyloid plaques before detectible symptoms, medics are pushing to treat Alzheimer’s disease as early as possible, says Malhotra. To try to detect the disease earlier, researchers are developing tests based on biomarker molecules found in patients’ blood. Options include two phosphorylated tau proteins, pTau181 and pTau217, which are good indicators of amyloid buildup, rather than tau buildup. Malhotra himself is helping run a clinical trial of pTau217 based at University College London. The proteins neurofilament light chain and glial fibrillary acidic protein, which normally help give brain cells shape, appear to predict neurodegeneration.
Everybody believes that this first generation of drugs is going to need to go into patients much earlier
‘Over the next five years or even less we will be able to take a set of bloods that will give us an idea of someone’s amyloid status, but also how much neuronal breakdown is taking place,’ Malhotra says. He adds that ‘it wouldn’t surprise me’ if blood biomarkers were soon felt equal to amyloid PET for diagnosing Alzheimer’s status.
Once diagnosed with presymptomatic Alzheimer’s, lifestyle factors may help people delay the onset of symptoms, Malhotra adds. ‘There seems to be some evidence that treating traditional cardiovascular risk factors, like high blood pressure, high cholesterol, may make a difference,’ he says. ‘My colleagues are setting up brain health clinics addressing relatively simple things like high blood pressure and environmental risk factors like air pollution, and hearing loss, which seems to be a risk factor too.’
Such tests may also see patients receiving antibody drugs sooner. ‘Everybody believes that this first generation of drugs is going to need to go into patients much earlier if you want to prevent the cell death,’ says Duff. ‘You’re going to have to get ahead of whatever the initiating neurotoxic event is.’
Pain
How does it work?
It’s easy to think of pain as one single thing – it feels that way – but it’s many. Usually, it’s picked up in our bodies when something stimulates specialised pain neurons, explains Gillian Burgess, head of research at Grünenthal, a pain-specialist pharmaceutical company based in Aachen, Germany. Known as nociceptive neurons, they can be activated to send pain signals to the brain by mechanical force or thermal energy. Chemical damage or other biochemical signals, such as substances released during inflammation, also activate nociceptive neurons. ‘Normally, that’s a good thing, because if you put your hand into a fire, you want to know about it,’ says Burgess.
Yet sometimes pain is less useful. Injury or disease in the nervous system itself can cause neuropathic pain that typically outlasts the triggering event, which can include stroke and nerve trauma. Burgess equates this type of pain to a disease. This overlaps somewhat with dysfunctional pain, chronic, widespread pain that occurs without stimulus, inflammation, or nervous system damage, typically caused by dysfunction of the nervous system amplifying sensory input.

What role does chemistry play?
To treat pain, we have a well-known arsenal of small-molecule drugs. Once inside our bodies, opioids like morphine and fentanyl interact with G-protein-coupled receptors (GPCRs), explains Susruta Majumdar from Washington University School of Medicine in St Louis, US. For example, the drugs first bind in a pocket in µ-opioid GPCRs on nerve cells, called the orthosteric site, shaped so that it normally accommodates natural painkilling peptide molecules. That binding changes the shape of the GPCR, making it couple to a G protein inside the cell, which triggers a protein called β-arrestin2 to move from the cell’s outer membrane to its nucleus. ‘From there to pain is this huge gap’ in understanding the biochemical processes involved, Majumdar says. ‘We’re still trying to figure out a lot of this.’
Opioids are ‘beautiful analgesics’, but come with the potential for addiction, and make breathing slow and shallow. Other painkillers include anti-inflammatory drugs like aspirin, which stops our bodies making prostaglandin molecules. Prostaglandin binding with GPCRs may make nociceptive neurons more sensitive. Other potential targets include GPCRs that naturally accommodate serotonin, γ-aminobutyric acid (GABA) and endocannabinoid molecules, but drugs that target them are less effective painkillers, Majumdar stresses. ‘You’re kind of limited so there is a necessity for coming up with new targets which will relieve pain,’ says Majumdar.
What have we learned in recent years?
It has become clear that experiments in animals to identify biochemical targets to treat pain are ‘often not translated through to success’, says Burgess. ‘Mice and rats are not humans, they have different physiology, and different molecular expression of proteins in their cells. I think these differences have been quite significant in our relative lack of success.’
Despite this, researchers are making good progress in developing a new family of pain drugs called sodium channel blockers. There are nine voltage-gated sodium channels found predominantly in nociceptive neurons, Burgess explains. Blocking sodium channels is the basis of the effectiveness of local anaesthetics, she says. ‘They block every single sodium channel, which is not necessarily what we want,’ Burgess adds. Some of them are found in the heart, brain or muscle, which prevents doctors giving the broad-spectrum blockers in high doses that affect the whole body.
‘The trick has been to try to isolate the ones that are expressed in in nociceptive neurons and hit them selectively,’ Burgess says. The best-known sodium channel target is NaV1.8. In early 2024 US company Vertex Pharmaceuticals announced positive phase III clinical trial results for its NaV1.8 blocker VX-548 in treating post-surgical pain.
What does the future hold?
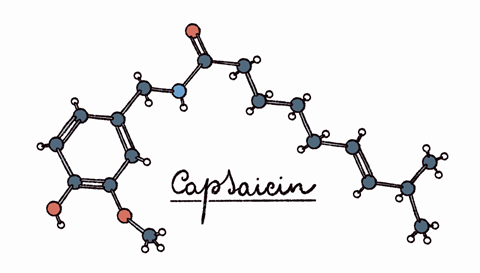
One of Grünenthal’s current products, Qutenza, is a patch containing capsaicin, the molecule that gives chilli peppers their heat. The company markets it to diabetics and people who have had shingles and are suffering from neuropathic pain. The drug activates the transient receptor potential cation channel subfamily V member 1 (TRPV1). Normally TRPV1 senses inflammatory molecules like prostaglandins, causing nociceptive neurons to register them as pain. TRPV1 also senses capsaicin, registering it as a burning sensation. However, in a high enough dose, capsaicin desensitises the TRPV1 channel, stopping it from registering any further painful sensation. The TRPV1 channels ‘open and they allow calcium to come into the very peripheral endings of these neurons, and they become defunctionalized’, Burgess explains. In essence, the chilli-flavour molecule uses up the nerves’ ability to signal pain.
The treatment is currently in clinical trials and the FDA has designated it a ‘breakthrough’
Now, Grünenthal is developing resiniferatoxin (RTX), which also targets TRPV1, but is ‘hundreds of thousands of times more potent’, explains Burgess. The aim is to give it as injections to provide long-lasting relief to people with osteoarthritis who are suffering from knee pain. The treatment is currently in clinical trials and the FDA has designated it a ‘breakthrough’, meaning that it will be assessed faster because it targets a serious condition.
Meanwhile, Majumdar’s team is working to design improved versions of existing pain drugs that don’t just bind to the orthosteric site in the GPCR that they target. This enables them to target GPCRs primarily responsible for pain relief, and not those responsible for side effects. Majumdar and his colleagues studied the μ-opioid receptor using cryo-electron microscopy, freezing the proteins and studying them in very high resolution. They worked out the detailed structure of the μ-opioid receptor, particularly where a sodium atom usually resides. They then synthesised a modified analogue of the highly potent opioid drug fentanyl that bound both the orthosteric site and the sodium binding site. In tests in mice, that drug increased rather than decreased breathing rates.
Memory
How does it work?
When living things learn and remember, many different biochemical processes happen on different timescales, explains Jelena Radulovic from Albert Einstein Medical College in New York and Aarhus University in Denmark. The fastest happens in just milliseconds in our neurons, of which humans have a staggering 85 billion, as ion channel proteins in their outer membranes open and close. When activated, the channels allow electrically charged ions of elements such as sodium, potassium and calcium to flow in and out. The ions rapidly flow through the neurons, becoming transient electrical signals. This happens repeatedly, with the ions measurably changing the voltage in the neuron in sudden spikes. The stronger the activating stimulus, the greater the neuron’s spiking frequency and voltage change.
The electrical signals soon arrive at junctions between neurons called synapses. If the activation stimulus has caused a strong enough signal, the neuron releases neurotransmitter signalling molecules, such as glutamate, into the synapse. The stronger the signal, the more synapses on a neuron can activate. This process happens on the millisecond timescale, Radulovic says. Another neuron on the synapse’s other side then has receptors to detect the glutamate molecules. The second neuron may then send its own electrical signal.
Activated neurons also change biochemically, using a complicated machinery of enzymes and proteins. The changes’ nature and duration depend on the strength of initial activation. Some neurons activate signalling pathways involving tagging proteins with phosphate groups. Kinase and phosphatase enzymes respectively add and remove these phosphate groups. The phosphates pass along a chain of different proteins, and may eventually reach the neuron’s nucleus, which houses its genes and controls its growth and metabolism. There they can change which genes the neuron expresses, altering the proteins it produces. This can maintain neuronal activity over minutes to a couple of hours.

In this time frame the synapses can also add more receptors, one aspect of the reshaping of neural connections known as plasticity. For example, if the second neuron activates after the first neuron, a complicated machinery of enzymes and proteins help add further glutamate receptors. Scientists refer to this as synaptic strengthening, or long-term potentiation (LTP). If the second neuron doesn’t activate after the first one, then some glutamate receptors might be removed, making activation after the first neuron less likely next time. That’s referred to as synaptic weakening, or long-term depression (LTD).
LTP and LTD can ‘maintain short-term memory’, says Radulovic, while changing gene expression forms long-term memories. Beyond phosphate-based genetic regulation, strong neuron-activating stimuli cause transcription factor proteins like Activating Protein-1 (AP-1) and Activating Transcription Factor (ATF) to make transient or persistent gene expression changes. ‘The genetic profile of the neuron is altered over long periods of time, in our studies over 21 days,’ Radulovic says. The genes involved include those that regulate synaptic connections and genes that affect the wider neuronal environment, known as the extracellular matrix.
This ‘bottom-up’ biochemical view of memory is relatively well-established. Yet, there is a lot of room for interpretation of how memory functions from a ‘top-down’ perspective of entire organisms and what happens in our brains. ‘When it comes to memory formation, and when it comes to learning, there is a lot of consensus in the field, but we also have different takes,’ Radulovic says.
What have we learned in recent years?
Over approximately the last decade, researchers have found that neurons activate together in groups, confirming a hypothesis proposed in 1949, says Clara Ortega-de San Luis from Trinity College Dublin, Ireland. The hypothesis refers to engrams, first proposed by German zoologist Richard Semon in 1921, small groups of cells that activate when we retrieve memories. In 1949, Canadian psychologist Donald Hebb proposed a central role for synapses in how engrams work in our brain.
We know that activation of 100 cells is sufficient to elicit the recall of memory
We now know that memory is recorded and recalled by sparse engrams of neurons, says Ortega. After a group of neurons is active during learning, they collectively undergo biochemical and physical changes to store information in a stable state and can be later reactivated, she says. ‘We know that activation of 100 cells is sufficient to elicit the recall of memory in some brain regions,’ she says.
Scientists like Ortega have developed engram labelling methodologies to investigate memory at the molecular and cellular levels. They rely on manipulating genes directly within live mice’s neurons that are thought to be part of an engram, using viruses that introduce genes that tell them to make proteins. Such cells can be made light-sensitive by engineering them to make a light sensitive protein called channelrhodopsin-2 . The researchers can stimulate these light-sensitive cells in mice to trigger a memory, for example making them freeze when recalling a fear memory. In this way the TCD team has found that postsynaptic density protein 95 (PSD-95) both influences synaptic plasticity and mediates engram connectivity patterns.
What other roles is chemistry playing?
In early 2024, Radulovic’s team found that long-term memory formation can cause DNA to break in some brain cells, most likely due to intense receptor activation. Afterwards, inflammatory response repairs the breakage and secures the memory.
The researchers trained mice to associate a small electrical shock with a new environment. When the animals were once again put into that environment, they remembered it with fear and froze in place. Using similar tools to those used to study engrams, they examined gene activity in neurons in the hippocampus, a brain area key to memory.
‘We got used to believing that memories are completely formed and that the long-term memories are already laid out within 24 hours,’ Radulovic says. ‘We found we had an early phase with kinase signalling, and gene expression within hours. Neurons showing such responses are typically viewed as engram neurons. However, then we had a period of maybe four to seven days when we had a very strong inflammatory response but in a different population of neurons.’
In these neurons, genetic responses were either disrupted or undetectable. Instead, they showed activation of the TLR9 protein, known to trigger inflammatory pathways in response to DNA fragments. Immune cells do something similar when defending against invading germs, but in this case, the nerve cells seemed to respond to their own DNA. Three weeks after training, the inflammatory genes were much less active. When the researchers deleted the gene encoding the TLR9 protein, the mice struggled to recall long-term memories about their training, freezing less often.
What does the future hold?
Thanks to the development of modern tools, we have learned a lot about memory, Radulovic says. However, ‘we scientists also need to be open to refining and even revising our concepts that relate to the neurobiology of memory if we are to make further progress’, she adds.
Ortega, meanwhile, wants more precise tools for labelling neurons. ‘Once we have that, we can much better understand how engram cells encode the memory,’ she says. ‘We know a lot, but we do not know enough.’
Andy Extance is a science writer based in Exeter, UK













No comments yet