
Developing drugs for treating rare diseases isn’t always financially viable. Clare Sansom looks at some recent success stories
Commercial drug discovery is exceptionally expensive, with the cost of taking a molecule ‘from bench to bedside’ estimated to be well in excess of a billion dollars. If a company is to recoup this financial investment while charging a price that health systems are prepared to pay, it needs to sell a large amount of every drug it develops.
The industry’s ideal disease is chronic, meaning patients stay on medication for many years. Ideally it would also primarily affect people in rich countries and have a large patient population. Patients diagnosed with rare diseases therefore have a significant disadvantage. This is not entirely a matter of economics: if there are only a few – or even a few thousand – patients to call on, setting up clinical trials with sufficient statistical power to detect an effect also becomes extremely difficult.
More people are affected by rare diseases than seems intuitively likely. In the EU, an estimated 30–40 million people suffer from a disease classed as rare. This disparity arises from the large number of such diseases. The definition of a ‘rare disease’ varies between jurisdictions: the US Food and Drug Administration (FDA) just considers prevalence (it must affect fewer than 200,000 individuals in that country) whereas the EU only includes diseases that are life-threatening or chronically debilitating. Whichever definition is used, the term encompasses over 5000 different conditions. About 80% of these have genetic or largely genetic causes, and many are diagnosed in infancy or early childhood.
Some countries have enacted so-called orphan drug legislation, offering scientific advice, reductions in fees and enhanced patent protection to companies developing medicines for these conditions. The European Medicines Agency (EMA) and FDA take a similar approach and work closely together. ‘We designate a medicinal product as orphan if its sponsors can prove that it is intended for the diagnosis, prevention or cure of a rare disease or one where there is insufficient incentive for drug development, and that it offers a significant benefit to patients,’ says Jordi Llinares Garcia, head of product development and scientific support at the EMA. Sponsors are usually, but not always, companies: ‘Individuals have occasionally applied for orphan drug designation and been successful,’ adds the EMA’s head of orphan medicines Kristina Larsson.
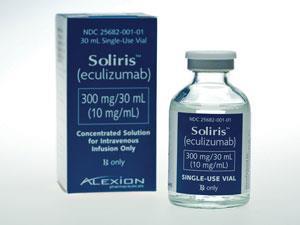
This legislation has enabled drugs to be developed for some of the very rarest diseases. Soliris (eculizumab) is a monoclonal antibody licensed for the treatment of two ultra-rare diseases involving blood clotting, paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome. This drug – which could potentially benefit about 200 patients in the UK – remains controversial because of its high cost, estimated to be about $400,000 (£260,000) per patient per year. This makes it one of the most expensive medicines in the world.
Commonest rare disease
Cystic fibrosis, which affects one in 2000–4000 Caucasian newborns, is one of the most common rare diseases. There are about 10,000 people living with the disease in the UK. It is inherited in a classic Mendelian recessive pattern. This means anyone who inherits a single faulty copy of the gene for the cystic fibrosis transmembrane conductance receptor (CFTR) will be healthy, but able to pass on the disease to their children, while anyone who inherits a faulty copy from both parents will have the disease. The protein produced from the CFTR gene forms a channel through which chloride ions pass through membranes and out of cells. These channels are important in producing mucus, sweat and digestive fluids. Cystic fibrosis patients, who lack working copies of this protein, suffer from a cluster of lung and digestive problems.
Not all CFTR mutations – and, therefore, not all CFTR patients – are equal, however. About half of all cystic fibrosis patients in the UK have two copies of a gene with the commonest mutation, in which one phenylalanine residue is missing. This produces an unstable and completely inactive protein, and severe disease. Patients whose CFTR protein contains a single mutated amino acid, in contrast, may produce a stable protein that functions inefficiently; the most common of these in the UK is the so-called Celtic mutation, G551D. Proteins with this mutation form a channel that forms on the cell surface as normal, but rarely opens.
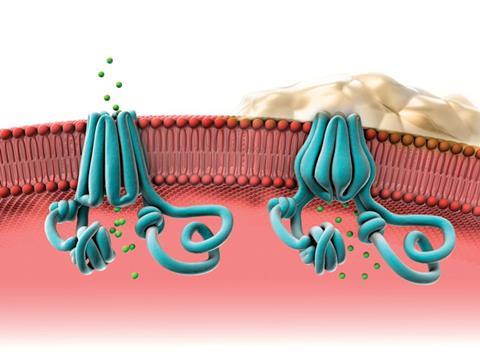
Until very recently, it was only possible to treat the symptoms of cystic fibrosis. The first drug to target the underlying defect, Kalydeco (ivacaftor) was developed by Vertex Pharmaceuticals (headquartered in Boston, US) and licensed in 2012. It is a fairly small and simple molecule – a dihydroquinoline carboxamide – that binds to the pore of the CFTR channel and changes its gating mechanism so the channel opens more often. It can, however, only treat patients with mutations like G551D where the defective protein is still expressed on the cell surface.
Jane Davies, a consultant in paediatric respiratory medicine at Imperial College London, UK, was involved in clinical trials of this drug in patients over six years old with at least one G551D allele. ‘We have shown the drug to be remarkably successful in improving our patients’ lung function and in maintaining that improvement for several years,’ she says. But there is, again, a controversy over its cost, which is about $300,000 per patient per year in the US (the UK cost has not been made public).
The development of Kalydeco is a significant breakthrough in cystic fibrosis, but one that can only hope to help a minority of patients with this disease. Vertex is currently investigating a combination of Kalydeco with lumacaftor in patients who have mutations leading to little-to-no CFTR protein on the cell surface, and has positive indications from Phase III trials. Gene therapy to repair or replace the faulty genes is also under investigation – the UK Cystic Fibrosis Gene Therapy Consortium recently completed a small Phase II clinical trial of the normal cystic fibrosis gene delivered into the cells using a liposome carrier. The data is still being analysed.
Boys’ network
Duchenne muscular dystrophy (DMD) has followed a similar pattern to cystic fibrosis, with dramatic progress being made treating a minority of patients with the disease. DMD only occurs in males, affecting about one in 3500 newborn boys. Boys with DMD inherit a single defective copy of the gene coding for dystrophin, a protein that forms part of a complex connecting muscle fibres to the surrounding extracellular matrix. They are generally wheelchair-bound by their teens and rarely live into their thirties.
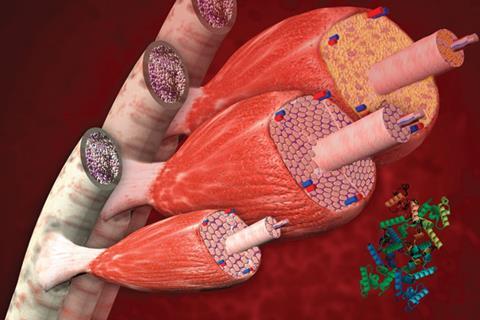
The gene coding for dystrophin is the largest in our genome – over 2.5 million base pairs long – and produces a protein with over 3500 amino acids. This gene, like most in the human genome, consists of a number of stretches of DNA that code for parts of the protein – the exons – interspersed with non-coding introns. Many disease-causing mutations in dystrophin involve the deletion of one or more of these exons, a process that can give rise to two different diseases. Most exon deletions cause the molecular machine that catalyses protein translation, the ribosome, to stop in its tracks along the messenger RNA so no protein is formed. In a minority of cases, however, the ribosome is able to skip past the deleted exon and continue translating the rest of the protein, so a shorter protein that retains some function is produced. Boys with these mutations have a much milder muscle-wasting condition, Becker muscular dystrophy, and can live into middle and even old age – although always with some disability.
Sarepta Therapeutics, based in Cambridge, US, is one of several companies developing methods to produce shortened but functional dystrophin protein where previously, due to genetic mutation, patients produced none. The technology, termed exon skipping, removes a section of mRNA following the genetic mutation, which enables the ribosome to continue translation from the start of the next exon, rather than stopping. This produces a shorter, partly functional protein, essentially transforming a case of DMD to a milder disease that might resemble Becker muscular dystrophy.
The company develops phosphorodiamidate morpholino oligomers (PMOs), which contain DNA bases connected to a backbone comprising six-membered morpholine rings linked by phosphorodiamidates (instead of the five-membered ribose rings connected by phosphodiester links in natural nucleic acids). Delivered to a cell, PMOs bind to targeted RNA and block access of cellular machinery to the RNA. This helps to repair defective RNA and restore translation of a desired protein.

The company’s lead product is eteplirsen, a PMO that binds specifically to a sequence in exon 51 of the dystrophin mRNA. Phase II clinical trials have already shown that the drug can restore dystrophin synthesis and prevent some muscular deterioration safely in pre-teenage and young teenage boys who are still able to walk. ‘Eteplirsen could treat about 13% of boys with DMD, a higher percentage than could be treated by a drug targeting any other exon,’ says Sarepta’s chief executive Chris Garabedian. ‘However, developing similar drugs based on the same approach would allow us to target many other mutations and address up to 80% of the DMD population.’
The disadvantage of this approach is that eteplirsen – like Kalydeco for cystic fibrosis – is only effective in a minority of patients. An alternative approach to potentially treat every DMD patient is to stimulate the production of another protein, utrophin, which has a similar function but is naturally synthesised by unborn babies.
Kay Davies from the Medical Research Council’s Functional Genomics Unit in Oxford, UK, and Steve Davies from the department of chemistry at the University of Oxford are taking this approach. ‘Steve and I started Summit Pharmaceuticals in 2003 to translate our many years of research into dystrophin and DMD into the clinic,’ says Davies. ‘There were fewer incentives for European companies to develop drugs for rare diseases then than there are now, and the best way forward seemed to be to start our own.’
Researchers at Summit used high throughput screening to select compounds that induced the production of utrophin and their lead molecule, SMTC1100, is now in clinical trials. ‘We don’t know this compound’s exact mechanism of action, but we do know that it works at the transcription level: that is, it induces the production of utrophin mRNA and thus of the protein,’ says Davies.
SMTC1100 completed a Phase I study with no serious toxicity issues and is now in Phase Ib clinical trials in a small number of boys with DMD. Some boys taking the drug have shown clinical improvement but the team has been challenged by the fact that patients respond in different ways. It appears that a boy’s diet affects the way the drug is absorbed and research is currently focusing on whether changing the way the drug is administered – it is currently delivered orally – would improve its absorption.
A sense of community
Despite the incentives offered by orphan drug legislation, companies are still reluctant to invest in early drug discovery programmes for rare diseases. The development of SMTC1100 highlights the way in which collaboration between companies and academic groups can help bridge this gap. And there is a third party that can have almost equal importance: the patients and their families. Some disease communities have their own national organisations to bring patients together to support research; patients with cystic fibrosis and muscular dystrophy are well represented by these. Some diseases, however, are so rare that their patient communities are too small to form effective support groups. Here, umbrella organisations, such as Eurordis– ‘the voice of rare disease patients in Europe’ – and Global Genes, based in the US but active worldwide, can be helpful.

Global Genes has collaborated with a unique patients’ organisation, PatientsLikeMe, to build a network of a million patients suffering from rare diseases. PatientsLikeMe was founded in 2004 by brothers James and Benjamin Heywood and a family friend, Jeff Cole, to support a third Heywood brother, Stephen, who had developed the rare disease amyotrophic lateral sclerosis (ALS). Their aim was to build up an online network of informed patients to discuss symptoms and treatments and pool their resources. Today, more than 300,000 patients have registered as users. ‘We have over 2000 different diseases represented, ranging from common ones such as breast cancer and Parkinson’s disease to some that have only ever been found in a few families,’ says Paul Wicks, vice president of innovation at PatientsLikeMe.
These networks of informed, connected patients are beginning to have an impact on clinical trials, Wicks explains. ‘In 2007 a group of patients with – coincidentally – ALS found a report of a small trial that suggested that lithium carbonate, which is used to treat bipolar disorder, might be able to control their symptoms. Within months, 160 patients from many countries had come together to obtain the drug off-label, monitored their own symptoms via PatientsLikeMe, and provided statistical proof that lithium was ineffective: four larger official trials proved the patients, rather than the scientists who conducted the first trial, to be correct.’
Genome sequencing
There are some patients who have no hope of a cure, or even of participation in a clinical trial, because they lack a diagnosis. But there is one almost sure way of tracking down even the most obscure disease: through sequencing a patient’s complete genome. Sequencing costs have dropped to a point when this is becoming feasible. One of the aims of the 100,000 Genomes project, launched in 2014 by Genomics England, is to sequence the complete genomes of thousands of patients with rare diseases and their close blood relatives. ‘This is a generic approach that can be applied to diagnose any rare inherited disease, and it will be available through the NHS in any region of the country,’ says Janet Thornton, director of the European Bioinformatics Institute (EBI) in Hinxton and chair of one of the working groups that set up the project.
It is likely that sequencing rare disease patients’ genomes will occasionally pick up defects in genes that have also been implicated in more common disorders, and, if the patient is lucky, a drug already licensed for the treatment of the common disease will help with the rare one. This synergy can also work the other way round. The Centre for Therapeutic Target Validation (CTTV) has been established at Hinxton – jointly by the EBI, the neighbouring Sanger Institute (where a third of the original human genome was sequenced) and pharmaceutical giant GlaxoSmithKline – to select, prioritise and study the best drug targets for a wide variety of diseases. ‘Sometimes studying the genetic cause of a rare disease will provide insights into appropriate targets for a common, multigenic one that shares the same pathway,’ says Ian Dunham, scientific director of the CTTV. ‘For example, some mutations in the gene NOD2 cause a rare inflammatory disease, Blau syndrome, while other variants have been associated with increased risk of inflammatory bowel diseases.’
These examples show how the prospects for patients with at least a few of the thousands of known rare diseases are being transformed, thanks to enlightened legislation, genomics and allied technologies and, not least, the involvement of the patients themselves. There is another type of disease, however, where the financial challenges of drug development are rather similar but prospects much less bright. These are the neglected diseases, such as schistosomiasis and Chagas disease, which affect many millions of people, almost all of whom are poor.
Clare Sansom is a science writer based in London, UK










No comments yet