Zeolites are important industrial catalysts, so why can’t chemists make them to order? Andrew Turley finds out

The preparation of catalytic zeolites can seem like a black art. You might read a paper and marvel at the obvious talent of the researchers, the elegance of the science and the tremendous impact of their findings – while at the same time wondering to what extent they genuinely understood what they were doing while they were doing it. Of course, the researchers themselves are not the problem. They know their area of science very well. The problem is simply that predicting zeolite function and synthesis is notoriously difficult.
But it is not impossible, and recent research suggests that the zeolite community is taking significant steps forward in this area. This would have a huge impact on the chemical industry, which relies on catalytic zeolites for a wide range of reactions crucial to modern society.
‘Serendipity plays a large part,’ says Mike Treacy, chair of the International Zeolite Association (IZA) structure commission and a physics professor at Arizona State University in the US. This is unfortunate because there are – at least in theory – an infinite number of framework types, something that is relatively easy to show in a thought experiment.
To get to grips with such an infinity, imagine three tetrahedra, each resting on one face with one vertex pointing up, their bases forming a triangle. Now, imagine a fourth tetrahedron, with the same orientation, on top of the three vertices. This gives you a tetrahedron of tetrahedra – a second generation tetrahedron. Finally, imagine swapping all the tetrahedra in it for second generation tetrahedra (see box below). The result would be a new and unique framework type, for which the same substitution can be made, and so on and so on.
In practice, of course, there are limits. As the cavities get larger, the zeolites get less robust, such that they slip out of their energy equilibrium with only the slightest provocation. ‘You’re going to have a huge cavity that’s one metre across, and there are only twelve bonds holding it together,’ explains Treacy. ‘If you as much as blow on it, you’ll break it. People have done these substitutions, but they’ve only gone as far as two.’
Building the catalogue
The number of achievable framework types is almost certainly rather less than infinite – but enormous, nonetheless. Treacy realised a long time ago that trial and error was not the ideal approach.
‘It is fair to say that, at present, the rate of discovery of zeolite frameworks that are uniquely useful to society is distressingly low,’ he wrote in a 2007 book chapter.1 ‘Given the huge economic benefits that are potentially available, a more rational approach to zeolite synthesis is needed. We need to know the frameworks in advance of synthesis, so that we can model their properties. The potentially useful frameworks then become synthetic targets and serendipity plays less of a role.’
Zeolite development would then be achieved in two distinct stages. The first would be design of the target; the second, design of an appropriate synthetic route to reach it.
Z is for zeolite
Zeolites are hydrated aluminosilicate minerals composed of linked alumina (AlO4) and silica (SiO4) tetrahedra. Cations of group one and two metals – commonly sodium, magnesium and potassium – sit in gaps in the structure and balance out the excess negative charge.
As a result, the elemental composition of zeolites is highly varied. Additionally, a wide range of structures are possible. The tetrahedra combine to form rings, cages and other characteristic substructures that create cavities of defined size and shape. These cavities play a key role in catalysis by making zeolites highly shape-selective. The selectivity can be based on the transition state of the target reaction or the exclusion of competing reactants. The catalysis is then facilitated by acid or metal centres within the framework.
Over the years, zeolites have become an extremely important class of industrial catalyst. They are widely used in the petrochemical sector to prepare hydrocarbons through cracking, alkylation and isomerisation.
Keeping track of such a diverse class of materials is a significant undertaking. The International Zeolite Association maintains an online database of zeolites that have been synthesised and fully characterised. Each entry corresponds to a framework type, identifiable by a unique three letter code. But this defines only the organisation of the component tetrahedra within the crystal structure. Each framework type refers to many zeolites that are structurally related but differ in their exact elemental composition.
The database currently contains 229 framework types. But new types are being added all the time: five in 2012, seven in 2013, 12 in 2014. In fact, the database is growing exponentially, doubling in volume every 16 years.
This would be tremendous for industry if every new framework type represented an advance in catalysis. But this is not the case. In general, the framework types in the database have been synthesised and characterised without explicit reference to functional properties. They were not, in other words, the result of any kind of targeting.
‘The targeted synthesis step is known to be a hard one,’ Treacy continued. ‘But there are no a priori reasons that it should be impossible. However, before we reach this stage, we need a database of potential frameworks whose physical and chemical properties are estimated.’ This database would be like a guidebook or catalogue for synthetic chemists, containing details of topology, unit cell dimensions and pore sizes. Treacy hopes it would be an interactive resource, allowing users to explore framework composition, visualise molecules passing in and out, and ideally would suggest template molecules for synthesis.
Treacy is part of a small community of researchers trying to map out the landscape of theoretical zeolites using computer simulations. He has created a set of 100,000 unique framework types that should be stable if they could be synthesised.2 His method is based around graphs made up from the connections between the atoms but without any distances or angles. Once he has the graphs, he imposes the necessary tetrahedral structures upon them. If they can be manipulated into sufficiently low energy conformations, they should be considered viable.
Elsewhere, other groups are chasing the same goal via subtly different means. Olaf Delgado Friedrich of the Australian National University and co-workers have used an approach in which pre-assembled polyhedra are tiled to fill the space. Meanwhile, David Earl and Michael Deem at Rice University in Houston, US, have produced a comparable set of theoretical zeolites via a different computational method that places greater emphasis on the positions of the atoms in the unit cell and less on the connections between them.
Choosing the target
These sets of theoretical zeolites are useful in that they provide clearly defined targets for synthesis. But the numbers involved are large. Picking a winner from 100,000 possibilities is not likely without some kind of guide.
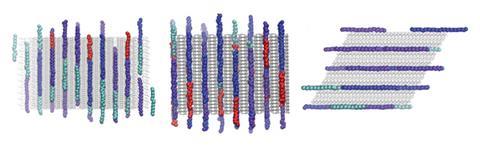
This is the problem that Ilja Siepmann and Peng Bai at the University of Minnesota in the US have been working on. In January, their team published details of a novel screening system designed to sort the wheat from the chaff.3 They focused on the hydroisomerisation of linear alkanes with 18–30 carbons. This is an important step in the de-waxing of certain oil fractions during the production of lubricants. The idea is to turn the linear alkanes into slightly branched ones of similar weight. This leads to lubricants that are suitable for lower temperature environments and thus more valuable.
To be suitable, the zeolite should have a high affinity for linear alkanes but a low affinity for branched alkanes. This ensures that the branched alkanes are ejected from the cavities quickly and reduces the likelihood that they will be cracked into smaller hydrocarbons of lower value. ‘There are a lot of back-and-forth reactions,’ explains Siepmann. ‘Linear gets converted to branched, and branched gets converted back to linear. Really, what is giving you the product selectivity is the dislike of the branched alkanes. Then whenever a branched alkane is produced in the catalytic reaction, it is getting pushed out of the zeolite and doesn’t have time to isomerise back to the linear one.’
Additionally, the zeolite should not be selective between different linear hydrocarbons. ‘In most applications, when you start from crude oil, you have a mixture of different chain lengths, and to some extent you do not want to give a preference to just convert the shorter or longer ones to the branched alkanes.’ Siepmann says. ‘You would like to convert them all.’
The researchers modelled the adsorption profiles of three linear alkanes and three branched alkanes. This required some very finely tuned algorithms for simulating molecular conformations and interactions, combined with a lot of computing power. Fortunately, the researchers had access to one of the most powerful supercomputers in the world, housed at Argonne National Laboratory near Chicago, US. Each simulation proceeds by placing the alkane within the zeolite in a stepwise manner. ‘We are starting with a first unit,’ says Siepmann. ‘And then this is looking around itself for empty space and putting the next unit where the empty space is.’
First, they validated their system using the framework types in the IZA database. Six of the top seven hits were for types described in patents for industrial hydroisomerisation of linear alkanes. This showed that their screening procedure, although based on adsorption properties, was able to capture the characteristics necessary for high-performing hydroisomerisation catalysts. They then moved on to a set of 300,000 theoretical types generated by Deem. ‘We found thousands that would perform better than any known material,’ says Siepmann.
The next step for the group is to try to synthesise some target zeolites so that their catalytic properties can be tested. This won’t be simple: synthesising zeolites is long and arduous. But as we learn more about how they form, we may be improving our aim.
Making the material

In June, the previously unknown structure of zeolite ZSM-25 was announced by an international team.4 This would be significant enough – ZSM-25 can selectively absorb carbon dioxide and its enormous structure has been a mystery for over 30 years – but there was more to the work than just structure determination. The team had been able to use the information acquired to rationalise the structures of two other zeolites, which they subsequently synthesised in a targeted fashion.
Paul Wright at the University of St Andrews in the UK became interested in ZSM-25 through the curious behaviour of another zeolite, Rho. The adsorption of carbon dioxide into zeolites is the focus of a lot of research because of the potential for its use in carbon capture and storage systems. Traditionally, the overall porosity of a zeolite is quickly tested by nitrogen adsorption at liquid nitrogen temperature: the more nitrogen adsorbed, the more porous the crystal structure and so the higher the expected capacity for carbon dioxide. But most cation forms of Rho don’t adsorb nitrogen readily, which might suggest it to be something of a dud. Wright found that, despite this, Rho adsorbed a relatively high amount of carbon dioxide. The anomaly, it turned out, was the result of metal cations acting like tiny trapdoors.
‘The structure is cages and windows,’ says Wright. ‘And there are cations in all the windows. No nitrogen goes in.’ But the carbon dioxide is able to coordinate to the cations, opening the doors slightly so that molecules can flow into the cavities behind.
After this, Wright turned his attention to other zeolites that had failed the nitrogen test. Could they also have potential as carbon dioxide adsorbents? ZSM-25 was one of these. Collaboration with Xiaodong Zou at the Stockholm University in Sweden revealed the structure via election diffraction. It was known that the structure of ZSM-25 contained certain symmetry elements also found in the structure of another zeolite, paulingite, which had already been fully characterised. This meant that the researchers could infer structural features of one from the other using strong reflections in the electron diffraction pattern.
They found that ZSM-25 was an expanded version of paulingite, with similar structural motifs. This led them to wonder whether the series could be extended into unknown territory through further expansion. Using the same ‘structural coding’ seen in paulingite and ZSM-25, they predicted new framework types in the same family. ‘Essentially, the method discovers certain characteristic features of the structure and then repeats them different numbers of times,’ says Wright.
Further collaboration, this time with a group in South Korea, was needed for the synthesis effort. The researchers at Postech in Pohang were able to synthesise the new framework types by adjusting the ratios of the metal cations to favour the formation of certain cages.
‘We knew that we wanted more of these cages, compared with ZSM-25,’ says Wright. ‘And one way to favour them would be to go to these divalent cations. So the same prep was used but with small quantities of added calcium and strontium.’
Wright believes the same principles might be applied to target other zeolites. ‘We think there are other families that this approach could be applied to, but we haven’t proved it yet.’
Catalysis by design?
What degree of control this work will enable remains to be seen. Certainly, novel catalytic zeolites that are tailor-made for specific reactions are still a long way off. But the field of catalysis in general may be going through something of a step-change in terms of control, according to Bruce Gates, a professor of chemical engineering at the University of California, Davis in the US.
There’s an enormous amount of trial and error in the history of zeolite preparation
‘Research strategies have gone well beyond resolving catalyst functions and elucidating catalytic sites by investigating simplified (model) catalysts,’ he wrote in an essay published in January.5 ‘Now catalyst preparation is morphing into catalyst synthesis, and flirtations with the notion of catalyst design are becoming serious.’
Gates sees the identification of base catalytic ‘functions’ as key. ‘You may say there’s a metallic function in a catalyst, and the metal might be nickel or it might be platinum or it could be something else,’ he says. ‘Let’s say I want to hydrogenate a double bond. Well, I’ve got a long list of metals I could choose from that would be active catalysts for that class of reaction. So there’s an element that you might say is related to design but it’s more primitive and you really shouldn’t use the word design.’
That said, Gates says that it is ‘entirely appropriate’ to use the word ‘synthesis’ in relation to ‘a number of catalysts that are less complex in structure and composition than the typical industrial solid catalyst’.
For Gates, zeolites may not yet be in the synthesis category but neither are they entirely random in nature. ‘There’s an enormous amount of trial and error in the history of zeolite preparation,’ he says. ‘Now, is it the monkeys at their typewriters? No. Because the people who are really good at this – and some people are extremely good at this – are being guided by good chemical judgements.’
Andrew Turley is a writer based in Cambridge, UK
References
1 Turning points in solid-state, materials and surface science, ed K D M Harris and P P Edwards, Royal Society of Chemistry, 2007, p208
2 M M J Treacy et al, Microporous Mesoporous Mater., 2004, 74, 121 (DOI: 10.1016/j.micromeso.2004.06.013)
3 P Bai et al, Nat. Commun., 2015, 6, 5912 (DOI: 10.1038/ncomms6912)
4 P Guo et al, Nature, 2015, 524, 74 (DOI: 10.1038/nature14575)
5 B C Gates, J. Catal., 2015, 328, 72 (DOI: 10.1016/j.jcat.2014.12.008)











No comments yet