π electron repulsion is the missing link in understanding conjugation
Scientists’ discovery that π electron repulsion is more important than previously thought might change our understanding of conjugation. The effect is behind peculiar irregularities in bond lengths and resonance energies of conjugated molecules that our textbooks cannot explain.
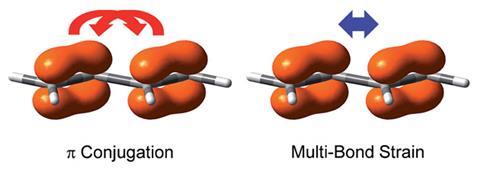
Conjugated molecules like 1,3-pentadiene have p electrons that are delocalised across several bonds. Conjugation lowers a molecule’s overall energy and makes it more stable. However, a few years ago, researchers from Long Island University, US, claimed that conjugation could actually destabilise some molecules. They calculated cyanogen (NC–CN) to be less stable than its non-conjugated analogue ethylenediamine (H2 NCH2–CH2 NH2). Many researchers have tried to disprove this, but so far, no one has been able to give a simple explanation for these apparently counterintuitive results.
Now, Yirong Mo from Western Michigan University, US, and colleagues discovered the missing factor to explain this strange destabilising effect: repulsion between π electrons. ‘I realised that all sides missed one key, the π–π repulsion,’ Mo says. Using a non-conjugated and a conjugated model system (B2 H4 and B4 H2) Mo and his team computationally measured repulsion between different types of bonds. They found that repulsion between conjugated p bonds was stronger than previously assumed – stronger even than the repulsion between adjacent s bonds, which pushes the molecule into the lowest energy conformation.
‘There has been overemphasis on conjugation, while the intramolecular multi-bond strain, which is at least equally important, has not received much attention. It’s about time to change the textbooks,’ comments Sason Shaik, an expert in computational chemistry at the Hebrew University, Jerusalem. Rainer Glaser, an expert in electronic structure theory at the University of Missouri, US, agrees. ‘This discussion is compelling and it should be and will have to be included in textbooks’.
Mo’s work can explain previously mysterious discrepancies between experimental and theoretical measurements, such as the longer carbon–nitrogen bond in nitrobenzene than in aniline; nitrobenzene’s aromatic ring and conjugated NO2 group repel each other through p–p repulsion, which doesn’t exist in aniline’s non-conjugated NH2 group.
Ángel Martín Pendás, a chemical bonding theory expert at the University of Oviedo, Spain, finds ‘the multi-bond strain concept a truly interesting proposal that deserves deeper scrutiny, and that will provide chemists with many hours of fun’.








No comments yet