
Manufacturer did not fully investigate reports of patients dying after auto-injector failures
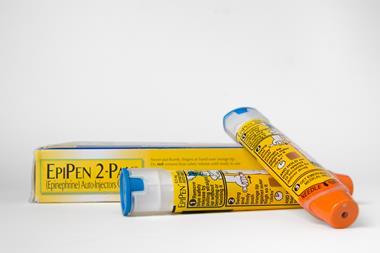
The US Food and Drug Administration (FDA) has formally warned Meridian Medical Technologies after the company failed to properly investigate reports of Epipen adrenaline injector malfunctions. Meridian has three weeks to notify the FDA of how it is addressing problems revealed in an inspection earlier this year.
The agency accuses Meridian, a Pfizer subsidiary, of ‘significant violations’ of good manufacturing practice requirements. For example, in February 2016, Meridian identified a fault in a batch of a crucial auto-injector component. Despite this, the company continued to manufacture Epipens using the component over the next eight months, while its supplier was investigating the issue.
The FDA also notes that Meridian received hundreds of complaints of Epipen products failing during ‘life-threatening emergencies, including some situations in which patients subsequently died’. However, the company did not thoroughly investigate these complaints, nor did it remove the potentially defective devices from the marketplace.
While 13 batches of Epipen products were eventually recalled from the US market, this only happened after the FDA’s inspection, and after ‘multiple discussions’ between the agency and Meridian.
Separately, Mylan – the distributor of Epipens – reached a $465 million (£360 million) settlement with US Department of Justice last month, over allegations it had knowingly misclassified the treatment to avoid paying rebates to government healthcare systems.














No comments yet