
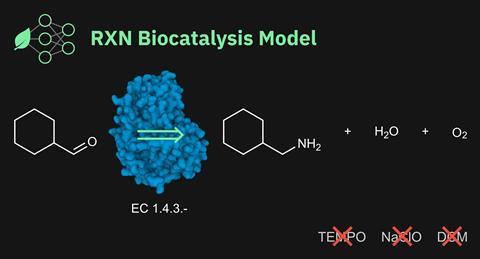
For the first time researchers have integrated enzymes into retrosynthetic chemical reaction planning using machine learning. Using data from four public biochemical reaction databases the team from IBM Research Europe in Switzerland taught its RXN for Chemistry tool both to predict reaction outcomes and plan chemical syntheses using enzymes.
This relatively small dataset of over 60,000 reactions enabled RXN to correctly predict the product from a given enzyme-mediated reaction on its first attempt nearly half the time. It could also plan out a potentially green retrosynthetic route using enzymes that would make a desired target chemical nearly 40% of the time. It was even able to correct some errors in the original datasets.
The key advance is proving that machine learning-driven reaction planning can extend to enzymes, says IBM Research Europe’s Daniel Probst. ‘To show that this actually works, even with this limited dataset, is the central point,’ he stresses.
Probst and his colleagues are seeking to make the chemical industry cleaner and more sustainable. Biochemical processes might be able to clean up traditional chemical processes that use toxic solvents, high temperatures and pressures, and produce lots of waste. Many companies already adopt enzymatic catalysis for faster, cleaner reactions, but available approaches are not yet as versatile as synthetic organic chemistry.
The RXN system uses IBM’s Molecular Transformer platform to predict the outcomes of reactions input by users. The program’s reaction planner can also suggest routes to desired molecules. Molecular Transformer uses natural language processing techniques also found in applications like automatic translation and voice assistants.
Extended chemical vocabulary
IBM Research Europe researchers adapted this artificial intelligence (AI) approach to extract reaction rules from chemistry patent data, so that Molecular Transformer ‘speaks chemistry’. ‘It learned how molecules look and interact,’ explained Probst, specifically learning to process a notation for chemical structure called Smiles.
Last year, IBM worked with the University of Bern to extend its vocabulary to enzymatic reactions. That approach trained neural networks with a text description of the enzymes, rather than its exact molecular structure. It could only predict reaction outcomes, not plan routes retrosynthetically.
Until recently, there was no automated way to plan enzymatic reactions. In 2021, however, William Finnigan and colleagues at the University of Manchester unveiled RetroBioCat. In the initial paper describing it, RetroBioCat included 99 hand-coded reactions. Defining each reaction rule manually might take a day, Probst estimates, but applying machine learning is faster. Initially adapting IBM Research Europe’s tool to enzymes took two to three months, says Probst, but then the data-driven method can scale up without defining new rules manually. ‘The question is whether it can find the same rules from the same data humans can,’ he adds.
Finnigan calls learning directly from databases rather than entering rules ‘very exciting’. Yet he knows of no good databases for using enzymes to make chemicals synthetically. ‘I believe the IBM tool is trained on several databases which have more of a focus on the function of enzymes in metabolism,’ Finnigan tells Chemistry World. As RetroBioCat focuses more directly on enzymatic synthesis the two approaches are complementary, Finnigan says. ‘At the end of the day, these tools should be seeking to augment a scientist’s abilities by posing new ideas and suggestions.’
IBM Research Europe is now working on understanding which aspects of the Smiles data the RXN model uses most to make its decisions. This may enable Probst and his colleagues to work out what rules form its biochemical knowledge, something that is hard to know when using machine learning – something often known as the ‘black box’ problem.
References
D Probst et al, Nat. Commun., 2022, DOI: 10.1038/s41467-022-28536-w
















No comments yet