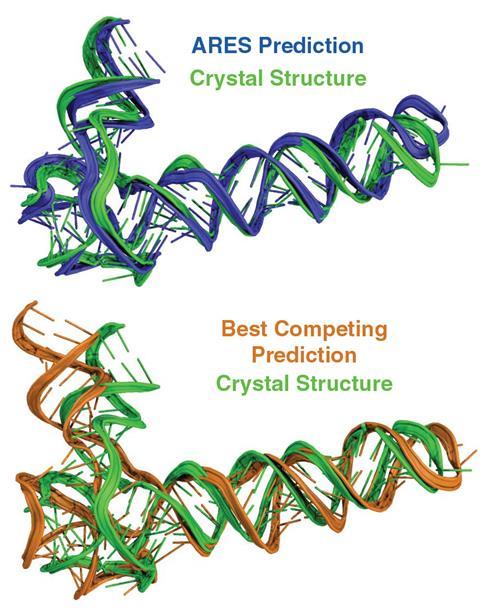
A team of biochemists and computer scientists has developed a new way to accurately predict the three-dimensional structures of RNA molecules, using an artificial intelligence system trained with a small number of known RNA shapes.
Experts have hailed the development as a significant improvement in the challenge of computationally predicting RNA structures, and say it could lead to a better understanding of RNA’s role in cellular functions and new therapeutic drugs.
Rhiju Das, an associate professor of biochemistry at Stanford University in California, says the new machine-learning system – called atomically rotationally equivariant scorer (Ares) – uses an ‘equivariant’ neural network to accurately distinguish the three-dimensional structure of an RNA molecule.
Das explains that the computational ‘neurons’ in equivariant neural networks do not solely use numbers for activation, like other types of neural network, but also vectors, tensors and other types of quantifiable objects. This allows Ares to assess the structural motifs of RNA molecules, such as different types of helices, ‘hairpins’ and stems – an approach called ‘geometric deep learning.’
Basic training
The researchers trained the Ares system on just 18 elaborate RNAs, whose structures were painstakingly determined experimentally. The system was then tested on much larger RNA structures listed on the RNA-Puzzles website, a decade-old scientific competition.
They used a version of the Rosetta molecular modelling software to generate more than 1500 different structure models for six solved RNAs from the website, while ensuring that at least 1% of them were ‘near native’ – meaning they corresponded closely to the true structure of the RNA.
They then used Ares to calculate a score for each of the model structures, as well as calculating their scores with the scoring functions of the Rosetta software, the ribonucleic acids statistical protocol (Rasp), and 3dRNAscore. The Ares system substantially outperformed the three other scoring functions: Ares included at least one of the ‘near native’ model structures in 81% of its 10 best-scoring models, compared with 48% for Rosetta, 48% for Rasp, and 33% for 3dRNAscore.
Ares also surpassed other scoring functions in tests with pools that included no ‘near native’ models. It also excelled at blind predictions in four rounds of the RNA-puzzles competition where the true structures of the RNAs were not yet known, giving the most accurate of the models submitted in every case.
‘It was a surprise that we were able to train the Ares network from so few training examples and then get state-of-the-art results on the RNA-Puzzles blind competition,’ Das says.
Playing catch up
The researchers wrote that scientific knowledge of RNA structure lags far behind that of protein structure, which benefits from artificial intelligence prediction systems like AlphaFold from the Google subsidiary DeepMind. These, by comparison, are often trained on huge datasets of thousands of structures.
‘The fraction of the human genome transcribed to RNA is approximately 30 times as large as that coding for proteins, but the number of available RNA structures is less than 1% of that for proteins,’ mainly because the structures of related RNAs are less likely to be known than they are for proteins and so cannot be used as templates, the researchers wrote.
They now hope the geometric deep learning approach pioneered by Ares will help stimulate research into RNA structures, although, so far, it only addresses one part of the process. ‘Our paper still relies on pools of models generated with a previous generation of the Rosetta software which didn’t make use of neural networks,’ Das says. ‘It would be wonderful to now generate the RNA 3D models themselves using tricks of geometric deep learning.’
And because Ares needs only atomic coordinates and chemical elements for its inputs, the same approach can be applied to other fields that involve three-dimensional chemical structure. Similar equivariant neural networks have been used successfully in recent research papers that use AlphaFold and Rosetta software, Das says.
Computational biologist Alex Bateman of the European Bioinformatics Institute, who was not involved in the study, notes that the prediction of RNA structures has lagged behind advances made in protein structure prediction enabled by AlphaFold. But ‘the development of Ares has shown a great step forward in the field and we are looking forward to getting access to these models’, he says.
He cautions that Ares still needs improvements in its accuracy. ‘Perhaps, inspired by the publication of the AlphaFold 2.0 method, we will see even better methods and models in the coming months and years,’ he says. ‘This is a very exciting time for RNA research.’
References
RJL Townshend et al, Science, 2021, 373, 1047 (DOI: 10.1126/science.abe5650)










No comments yet