
Scientific fields from drug discovery to plastic recycling have been transformed after artificial intelligence (AI)-based protein structure prediction researchers shared an enormous dataset and opened up their methods. On 22 July, London, UK-based Google offshoot DeepMind and the European Molecular Biology Laboratory (EMBL) released 350,000 protein structures computed by its AlphaFold system. These include all 20,000 proteins from the human proteome and will be followed by over 100 million more.
The AlphaFold team stresses that this is ‘almost every sequenced protein known to science’. Current estimates indicate that 200 million or more protein structures exist in nature. They’re molecular machines made of single long strands of amino acid building blocks that fold up into unique 3D shapes. Knowing those shapes is vital for understanding how the proteins work.
Demis Hassabis, the founder and chief executive officer of DeepMind, described this as ‘the most significant contribution AI has made to advancing scientific knowledge to date’. Hassabis said that it will have applications that range from ‘drug discovery, to protein design, to disease understanding and to enzyme design’. AlphaFold has already helped engineer faster enzymes for recycling single use plastics, he added.
‘For me, this dataset is rather like the human genome,’ comments Ewan Birney, deputy director general of EMBL, noting that the resource will enable previously impossible science. ‘I’m very, very excited to start walking down that road.’
DeepMind’s announcement came after both the AlphaFold team1 and David Baker’s RoseTTAFold group2 at the University of Washington in Seattle, US, described their systems and made their code openly available on 15 July. In December 2020, AlphaFold won a protein structure prediction competition known as the Critical Assessment of Structure Prediction (Casp14) challenge. However, DeepMind had not revealed how its system worked.
Details delivered
The RoseTTAFold team sought to develop its own protein prediction tool, describing how they built on just five hints picked up from DeepMind in their study. ‘It’s like you heard about a great painting and tried to copy it without any details, like which colour or what approach they used,’ says University of Washington computational chemist Minkyung Baek, who led the RoseTTAFold project.
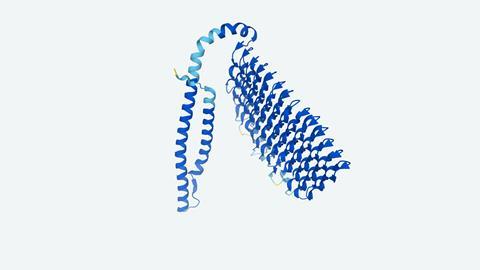
The two teams’ architectures both start by gathering protein sequence data from genetic information via multiple sequence alignment (MSA), which is a process that collects all known sequence that are evolutionarily related to a protein of interest.
DeepMind’s AI feeds both the MSA and a table of pairing information into a neural network dubbed Evoformer, organising the data and grouping similar proteins. It also inputs related known protein structures as templates to learn from. The system then passes Evoformer’s outputs into a second neural network, the structure module, to predict the target protein’s 3D shape. All the while, it estimates the predicted structure’s accuracy compared to real protein structures. AlphaFold continually feeds the outputs of Evoformer and the structure model back into the start of process until those estimates no longer improve.
In contrast, RoseTTAFold relies on three neural networks, organised into separate tracks. The first continually refines an MSA. The second does the same for information about pair interactions derived from the MSA and known template protein structures. These run in parallel, each feeding back to the other to improve their predictions. After initially optimising predictions in this way, RoseTTAFold then adds a third structure-generation track, which also feeds back to the first two tracks. That allows the entire network to collectively reason about relationships between sequences, pairwise distances and 3D shapes.
Both AlphaFold and RoseTTAFold can predict the structure of a protein with around 400 amino acids in just a few minutes.
How good are these tools?
Simon Erlendsson, a biochemist at the UK Medical Research Council Laboratory of Molecular Biology in Cambridge, says that the research world is ‘boiling’ with talk of the new tools and their predictions. ‘AlphaFold and RoseTTAFold represent tremendous breakthroughs and will undoubtedly guide structural biology and sophisticated protein design for many years to come,’ he says.
Erlendsson studies brain proteins whose structure researchers haven’t completely figured out, like Arc, which helps regulate memory. He notes that AlphaFold’s prediction suggests a way that Arc might interact with itself that nobody has seen before. ‘I’m actually a little bit embarrassed that I didn’t see this in the first place,’ Erlendsson says. ‘But maybe it’s not there, right?’
He raises a crucial question about how good these protein predictions really are. At Casp14, AlphaFold’s median accuracy for protein backbones was within the width of a carbon atom. However, John Jumper, AlphaFold lead at DeepMind, notes that the system works best when three conditions are met. There must be good information on a protein’s sequence, at least 30 related sequences whose structures are unknown, and some related sequences with known structures. Accuracy ‘drops substantially’ when these inputs are weaker, he says.
Another potential problem is that proteins can adopt different forms, and that these prediction models may provide less interesting inactive structures. For example, G-protein coupled receptors – the ‘locks’ on our cells that trigger changes in response to chemical keys – have active and inactive forms. To ensure that RoseTTAFold outputs the active form in that case, Baek explains that she only let it learn from active forms of known template protein structures.
Despite such potential issues, Kathryn Tunyasuvunakool, a senior research scientist at DeepMind, says that AlphaFold can already help physical structure studies. ‘Sometimes when someone has obtained an x-ray crystallography dataset they can’t immediately determine the atomic structure of the protein from that,’ she explains. ‘It helps to actually have a good initial guess at the structure.’
While these new techniques hold great promise, Baker doesn’t consider them to have entirely answered the protein folding problem. ‘If by the protein folding problem you mean predicting the structure of a protein from its primary sequence, then these methods are close to a solution,’ he says. ‘However, they are essentially doing pattern recognition. They do not describe the process by which a protein goes from an extended chain to a folded structure.’
References
1 J Jumper et al, Nature, 2021, DOI: 10.1038/s41586-021-03819-2
2 M Baek et al, Science, 2021, DOI: 10.1126/science.abj8754











No comments yet