
A neural network trained on a small dataset allows researchers to accurately predict the enantioselectivity of asymmetric catalytic reactions. The system, developed by researchers in the US, enabled the design of new ligands that could boost enantioselectivity in a cross-coupling reaction.
Screening ligands is essential for optimising the selectivity of catalytic reactions, but the process is time and resource intensive. Approaches using density functional theory (DFT) are sometimes used to model enantioselectivity to try to speed up the ligand selection process, but navigating narrow energetic differences to get an accurate picture of how well a chiral catalyst will work is challenging. While machine learning models could potentially enhance these predictions, they traditionally require large datasets.
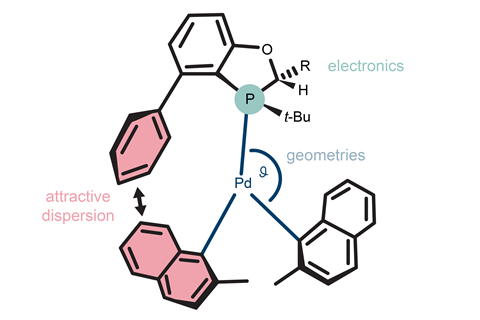
Now, researchers from Yale University and Boehringer Ingelheim Pharmaceuticals have used a neural network to harness the full potential of a small dataset of DFT-calculated molecular features for a Negishi cross-coupling reaction. The neural network, which was trained with data describing the transition state free energies, geometries and electronic features of just 17 phosphorus ligands, was able to provide a highly accurate model for enantioselectivity. The method is also the first to consider attractive dispersions as part of a neural network that describes reaction efficiency.
The most important catalyst features influencing the model’s performance were the valence electron population at one particular carbon centre, deviation from planarity by the metal centre and the bond angle at a ring junction adjacent to the phosphorus atom.
The neural network enabled the team to identify a new ligand with greater enantioselectivity than any of the input ligands, with the predicted selectivity in good agreement with experimental results. The researchers suggest that the technology could be implemented for a wider range of reactions – particularly those for which experimental data is limited.
‘Large language models, such as ChatGPT, are trained on a vast amount of text. There is no equivalent dataset for chemistry. Even PubChem, with more than a hundred million molecules, is tiny in comparison.’ notes Jonathan Goodman, an expert in chemical informatics from the University of Cambridge, UK, who was not involved in the project. ‘A different approach is needed to train a neural network to be useful for chemistry. This [work] shows that it is possible to combine experimental data, DFT calculations and human insights in order to generate an effective model for selectivity. This is likely to inspire others.’
References
A E Cuomo et al, ACS Cent. Sci., 2023, DOI: 10.1021/acscentsci.3c00512














No comments yet