Study confirms mechanism of sensitisation-initiated electron transfer reaction following extended debate
Researchers have provided spectroscopic evidence for the mechanism of a two-photon photoredox process known as sensitisation-initiated electron transfer, or SenI-ET.1 They applied transient absorption and emission spectroscopy to an iridium-based photosensitiser–catalyst couple, which allowed them to identify the active catalytic species – a short-lived pyrenyl radical anion – for the first time. The researchers hope their work will inform the design of new photoredox systems based on multi-photon excitation, with potential applications in synthetic organic photochemistry and solar fuel conversion.
Typical photocatalysts rely on excitation with a single photon to produce a reactive intermediate that can trigger chemical transformations. Recent years have seen the development of multi-photon excitation strategies, which can produce highly reactive intermediates otherwise inaccessible via the energy input of a single photon. While multi-photon systems have made thermodynamically demanding reactions more accessible, the multitude of potential reaction pathways and the short-lived nature of radical intermediates has made mechanistic analysis challenging. Scientists hope that uncovering the reaction pathways of multi-photon processes will permit the design of more efficient photoredox catalysts in the future.
A 2017 study2 by Burkhard König and co-workers at the University of Regensburg in Germany sparked debate by proposing the biphotonic photoredox process known as SenI-ET, which combines energy and electron transfer steps to generate the key catalytic species. In the study, the group applied a [Ru(bpy)3]2+/pyrene/DIPEA photoredox system to the reductive dehalogenation of aryl halides and proposed a mechanism for forming the presumed catalytic species, a pyrenyl radical anion (Py˙−). A later study,3 led by Paola Ceroni and Vincenzo Balzani at the University of Bologna, disputed the proposed mechanism and the debate continued. Even a 2020 intervention by Evan Moore and co-workers at the University of Queensland failed to resolve things, with their laser spectroscopy approach yielding yet another dominant mechanism.4 Amid all this, experimental evidence for the key Py˙− species remained elusive.
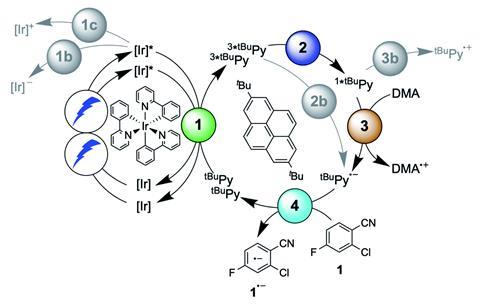
Now, a team led by Oliver Wenger at the University of Basel in Switzerland, has applied transient absorption and emission spectroscopy methods to a fac-[Ir(ppy)3]/tBuPy system, aiming to confirm the dominant reaction pathway in the SenI-ET process. By carefully selecting the photosensitiser–catalyst couple, the group were able to suppress the initial electron transfer step, thus dramatically simplifying the mechanistic analysis. Further still, the group’s system was designed to disfavour a side reaction causing rapid reduction of the key Py˙− species, enabling a full spectroscopic characterisation of each reaction intermediate for the first time. Using this information, the group concluded that the reaction proceeds via a sensitised triplet–triplet annihilation upconversion pathway, as originally postulated by Ceroni and Balzani. With the mechanism finally confirmed, Wenger’s team carried out hydrodehalogenation as well as detosylation and pinacol coupling reactions to demonstrate the applicability of their system to preparative-scale photoredox catalysis.
Ceroni is impressed with the mechanistic focus of the study. ‘Elucidation of the mechanism is of the utmost importance to improve photocatalyst design and efficiencies. Up to now, most papers report tentative mechanisms without clear experimental proof, so that developments are mostly based on trial and error approaches. [This is] an astonishingly detailed study… and one of the rare examples of a paper strongly focused on the photochemical mechanism,’ she tells Chemistry World.
For Wenger, future work will aim to extend the catalytic potential of the group’s approach. ‘Previous studies exploited upconverted singlet-excited states directly for photocatalysis. These singlet-excited states are typically very short-lived, which limits their kinetic reactivity. In the future, we plan to take advantage of the concept demonstrated in our latest study, which consists of quenching the short-lived singlet-excited states by a sacrificial reductant to obtain a radical anion that is not only much longer lived, but can be thermodynamically more reactive.’
References
1 F Glaser, C Kerzig and OS Wenger, Chem. Sci., 2021, DOI: 10.1039/d1sc02085d (This article is open access.)
2 I Ghosh, RS Shaikh and B König, Angew. Chem., Int. Ed., 2017, 56, 8544 (DOI: 10.1002/anie.201703004)
3 M Marchini et al, Angew. Chem., Int. Ed., 2017, 56, 12820 (DOI: 10.1002/anie.201706217)
4 M S Coles et al, Angew. Chem., Int. Ed., 2020, 59, 9522 (DOI: 10.1002/anie.201916359)











No comments yet