A rare example of a [6+2]-cycloaddition
I wrote the other month about my love of cycloadditions (cycloaddiction, if you will), and we’ll see one take centre stage again today. But before we get into that, I need to get something off my chest about the way we name these transformations. Take the most famous, textbook example – the Diels–Alder reaction. In its simplest form, a diene reacts with an alkene to form a cyclohexene ring. In the language of the Nobel-prize-winning system laid down by RB Woodward and Roald Hoffman, that’s a [4+2]-cycloaddition because of the number of electrons in the reacting partners – four in the diene and two in the alkene.
Simple, right? However, before this electron-centric convention was introduced in the 1960s, such reactions were instead named by the number of atoms in the reacting parts of the molecules. This doesn’t change anything for the venerable Diels–Alder (it’s a 4+2 in either case), but there are examples where the number of reacting atoms and the number of electrons involved aren’t the same.
To avoid potential confusion, Iupac (the arbiter of chemical convention) tells us to use regular curved brackets when talking about the number of atoms in a cycloaddition, reserving the square ones for conversations about electrons. Unfortunately, this point of pericyclic parenthetical pedantry is often missed, and the literature is rife with rule-breaking (often abetted by the word ‘formal’). This caused me a great deal of confusion in my early years, and I’d advise you not to infer too much from an author’s choice of bracket.
OK – that was a little more educational than this column typically strives to be, but if I’ve saved a few readers from hunting for missing electrons, it will have been worth it.
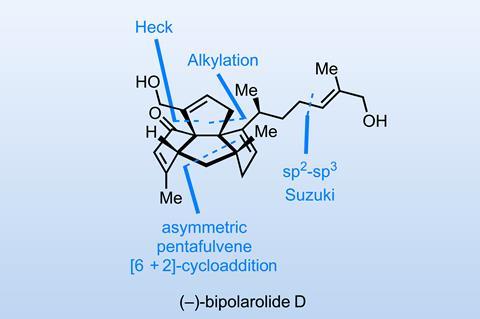
Now, if you read a dozen randomly chosen total syntheses, I’d bet good money that you’ll see several Diels–Alder reactions. You might also sight a less-common [2+2] or [3+2] and, if you’re very lucky, even catch a glimpse of something as exotic as a [5+2] or [4+3]-cycloaddition. But a recent synthesis of bipolarolide D from Zhaohong Lu’s group at Xiamen University in China showcases something I don’t think I’ve seen en route to a natural product before – a [6+2]-cycloaddition reaction!
The starting material is a pentafulvene, a class of molecules far more common in organometallic chemistry or molecular electronics than organic synthesis. With the addition of a little Hayashi–Jørgensen catalyst, an enamine is formed (figure 1). This then serves as the two-electron component in the ensuing asymmetric [6+2]-cycloaddition, with enantioinduction provided by the proline ring and the product formed in good yield and excellent enantiomeric excess.

Next, a challenging regioselective alkylation begins the installation of the top ring. Surprisingly, strong bases are no good here, and it’s tert-butoxide, buffered with just the right amount of tert-butanol additive, that does the trick. The yield is also buoyed by the addition of copper powder, which helps protect the fragile-looking electrophile and product from decomposition (commercial allyl iodide is often stabilised with copper).

Next, a Heck cyclisation snaps the final ring shut, and the η3-allylpalladium intermediate is trapped with acetate, which the team immediately hydrolyses to the alcohol (figure 2). The reaction is remarkably high-yielding and selective, given multiple possible reaction sites here. From here, a neat rhodium-mediated redox-neutral reshuffle oxidises the new alcohol and transfers the borrowed hydrogen atoms to the unwanted olefin in the central ring, with selectivity for this olefin presumably driven by strain at the bridgehead. Look Ma – no reductant! Finally, a sterically-driven regio- and diastereoselective Grignard addition completes the carbon skeleton of the target.
From here, all that remains to do is make some redox adjustments and bolt on the terpenoid sidechain. Congratulations to the team on a highly efficient synthesis of this dense polycyclic natural product!
References
1 Z Lu et al, J. Am. Chem. Soc., 2024, DOI: 10.1021/jacs.4c04059









No comments yet