A non-obvious combination unleashes the power of electrocyclisation
Long-time readers of this column will know that I love a cycloaddition – I’ve probably run hundreds of them over my career as an organic chemist. There was even a two-hour period immediately before my PhD viva where I could credibly claim to understand the fiendishly complex Woodward–Hoffman rules that were used to rationalise their outcomes in the pencil-and-paper days before computational chemistry. However, I have far less experience with their rarer cousin, the electrocyclisation.
On the surface, these reactions are pretty simple: electrons dance around a cyclic transition state, resulting in a double bond lost and a single bond gained. Although not obvious at a glance, the stereochemical outcome is actually highly predictable, providing your grasp of molecular orbital theory is strong and you have a spare 15 minutes. But if you ask me, predicting the product is the easy part. The greater challenge from a total synthesis point of view is spotting where these reactions fit into a route and can actually add value. Most people, myself included, tend to retrosynthesise by breaking bonds, not moving them. And after the effort of setting up an elaborate substrate to rearrange with often a minimal jump in complexity, you’re left with the feeling that the juice wasn’t worth the squeeze.
During my postdoctoral studies in the US, the research group I was in would periodically split into teams to tackle a ‘molecule of the month’. The goal was to hone our retrosynthesis skills by devising the best possible total synthesis of some daunting natural product and then defend this proposal to a room of increasingly inebriated and combative chemists at one of our 8pm Monday night group meetings. Free beer was provided; the winner was chosen by a vote and the loser was anyone who had serious work to do the next morning. Some of those nights are a little hazy, but I’m pretty sure that in my two years of participating in these competitions, I never saw an electrocyclisation proposed. And that probably means something.
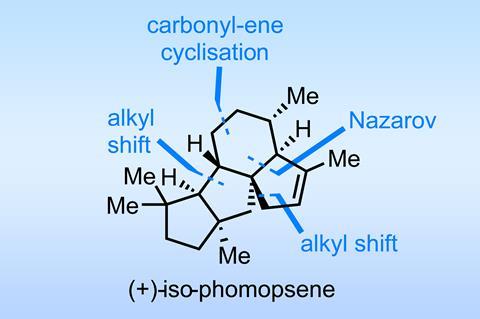
A great example of a recent total synthesis that hinges on a couple of well-chosen (but well-hidden) electrocyclisations is the route taken to the phomopsene diterpenes by Yong-Qiang Tu of Shanghai Jiao Tong University and Lanzhou University, China, and co-workers.1
The route more-or-less begins with a Nazarov cyclisation/ring expansion cascade, an unbelievable disconnection that’s about as obvious as a black cat in a coal cellar. This neat little electrocyclisation, followed by a brace of alkyl shifts, stereospecifically establishes three rings and two contiguous stereocentres (figure 1) – and from a dienone precursor starting material that’s actually pretty trivial to make, at least racemically.

A couple of steps on, it’s time for a third ring expansion. A nifty one-pot sequence involves the Beckmann fragmentation of an oxime, followed by diisobutylaluminium hydride (DIBAL) reduction of the resulting nitrile to the aldehyde and finally a carbonyl-ene reaction (figure 2). The fact that the electrocyclisation is triggered by neutral alumina – about as innocuous a solid as is possible – shows just how well the system is set up for this reaction, which proceeds with excellent diastereoselectivity.

Overall, this formal C–H functionalisation on an ordinary-looking methyl is an unusual and creative twist for a functional group often thought of pretty inert, especially when found on a quaternary centre. Tying the vision of this rearrangement sequence to the Nazarov product shows a masterful level of synthetic planning, deploying two rarely seen and non-obvious reactions to great effect.
I highly recommend reading the full synthesis, particularly for the dizzying jump in complexity over the first seven steps, which would make for a fiendish problem for a graduate organic chemistry class!
References
1 Y-Q Tu et al, J. Am. Chem. Soc., 2023, DOI: 10.1021/jacs.3c07044











No comments yet