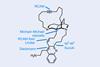
(+)-Njaoamine I
Ringing in the advantages of alkyne methathesis
When I think about the new chemical reactions discovered in my lifetime, they’ve been less ‘Eureka!’ and more grind. As organic chemistry prepares to close out its second century, we’re not tripping over new reactions the way that we used to, and those we do find tend to be far more complex and in need of fine-tuning than those we’re taught in school. The gap between such methodologies appearing in the pages of a journal and chemists trusting them enough to plan their use 15 steps into a total synthesis is often measured in years – if not decades.
One reaction that’s been hovering on the edge of relevance for a few years is alkyne metathesis, which is generally viewed as a poor cousin to the Nobel-winning alkene version, despite being discovered around the same time. Unfortunately, although the ability to chop-and-change triple bonds at will sounds cool, the reality has generally been less appealing. The necessary catalysts are far less well-developed (read: generally finicky), and there’s also the cost of preparing the alkyne starting materials, which take more steps and effort than simple alkenes. However, ring-closing alkyne metathesis (RCAM) in particular has at least two great strengths. The first – and most obvious – is that it produces cyclic alkynes. True, we don’t need to make these often, but there actually aren’t many appealing options to access them. Secondly, RCAM sidesteps the great bugbear of alkene metathesis – controlling alkene geometry. As powerful and easy as alkene metathesis now is, it’s often bedevilled by mixtures of products, which are famously difficult to separate. RCAM can solve this problem as many reliable methods exist to convert the alkyne products to either geometry of alkene (albeit at the cost of a few more steps).