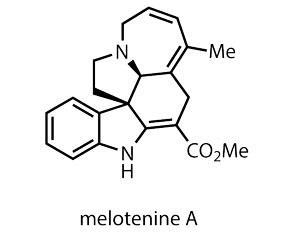
Deftly demonstrating this utility is a fresh synthesis of a recently discovered indole alkaloid. Bearing only two stereogenic centres, at first examination melotenine A doesn’t appear to be the most complex of targets. However, the pentacyclic arrangement with the stereocentres at the core arouses the prospective chemist’s imagination. Identified only three years ago, this alkaloid immediately stimulated interest after showing promise in the familiar cancer cell-line smackdown.1 The isolation team suggested a relatively simple biogenesis starting from structurally related tabersonine, so the synthetic challenge was open.
A team led by Rodrigo Andrade of Temple University in Philadelphia, US, rejected this potential route and began from scratch.2 Starting from the commercially available N-tosyl indole-3-carboxaldehyde, the team immediately took advantage of a simple yet relatively exotic chiral sulfinamide. Bearing three substituents and a lone pair of electrons, the sulfinamide can act as a chiral ammonia equivalent. This unusual feature has brought it fame and fortune (well, not personally) as both a substrate and, as in this case, a chiral auxiliary. Condensing the sulfinamide with the indole aldehyde forms an imine, and the chiral sulfur group can then direct a highly diastereoselective Barbier coupling reaction with allyl bromide (figure 1).
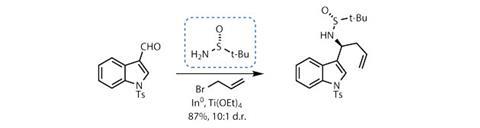
Having performed its role admirably, the sulfur group leaves the stage, revealing an unsubstituted amine. This is immediately augmented with another side chain by reductive amination, then protected with a tert-butoxycarbonyl (Boc) group. The terminal olefin is also extended by metathesis with a little methyl acrylate and Hoveyda–Grubbs second generation catalyst.
With these simple modifications complete, it’s time to build some rings. The US team does this in style. Taking inspiration from Jonathan Ellman of Yale University, US,3 they use a Mitsunobu reaction to build a pair of rings in a single process. The indole enamine closes down onto the activated alcohol of the amine side chain to form the pyrrolidine ring. Adding base and heating gently then closes down a fourth ring (figure 2).
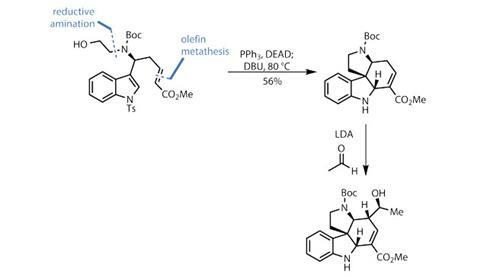
This relatively simple process delivers the target tetracycle in an impressive 56% yield, and as a single stereoisomer. Adding a slightly beefier base and some acetaldehyde then promotes a vinylogous aldol reaction, adding a two-carbon unit in both high yield and diastereoselectivity. A few steps further on, the team uses this exocyclic ketone to construct the final seven-membered ring.
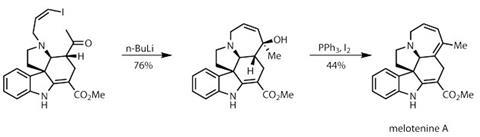
Before this final ring formation, the team must move the double bond of the a,ß-unsaturated ester one place round the six-membered ring. They do this by first reducing the olefin, and then oxidising it back into the required position. They then deprotect and alkylate the pyrrolidine with bromo-1-iodopropene, completing the carbon count in preparation for the final cyclisation. To build the tetrahydroazepine ring, lithium–halogen exchange with the vinyl iodide creates an intramolecular nucleophile that reacts with the recently installed ketone.4 Dehydrating the resultant alcohol completes the target (figure 3).












No comments yet