When amides get out of shape, a whole new world of asymmetric aldol reactions opens up, says Karl Collins
When I think of asymmetric aldol reactions, what immediately springs to mind is using boron to control enolate geometry and chiral auxiliaries to impart facial selectivity in the addition step. If I try a little harder, Mukaiyama aldol chemistry using pre-formed silyl enol ethers, with facial control from chiral Lewis acids, emerges from the deeper recesses of my memory. However, it is rare that catalytic enantioselective aldol chemistry makes an appearance.
Masakatsu Shibasaki of Tokyo University, Japan, developed the first catalytic enantioselective direct aldol reaction – one which does not employ preformed enolates – in 1999.1 While significant effort has been expended since then, the fact that auxiliary-based reactions still broadly dominate the field indicates that a lot of progress is still to be made.
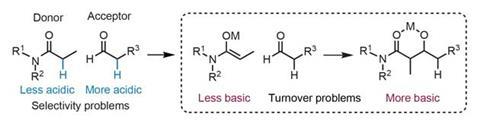
Catalytic aldol reactions face two major challenges (figure 1): firstly, the aldol donor must be selectively transformed into the desired enolate, in the presence of a typically more acidic aldehyde acceptor. This broadly limits the range of aldol donors to relatively acidic aldehydes and ketones, with less-acidic carbonyls – particularly amides – seldom appearing. Second, the reaction products are more basic than the starting materials. The product strongly coordinates the Lewis acid catalyst, trapping it and inhibiting turnover. Moreover, the reverse reaction must be inhibited once the product is released.
An intuitive approach to selectively forming amide-derived enolates would be to make them more acidic by adding electron-withdrawing groups. But a team, led by Shibasaki and Naoya Kumagai, has developed an intriguing alternative.2 Rather than exploiting the electronic properties of the amide to enhance the acidity of the enolisable proton, they induce conformational constraints on the aldol donor using an external Lewis acid. This renders the latent amide enolate suitably acidic to allow for selective deprotonation in the presence of an aldehyde acceptor.
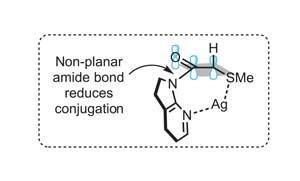
Using alpha-sulfanyl 7-azaindolinyl amide as an aldol donor, they propose that introducing AgBF4 forms a seven-membered bidentate chelate structure (figure 2). This both tilts the indolino group, reducing the amide conjugation, and aligns the appropriate C–H bond with the p-orbital of the carbonyl to further enhance its acidity. Using this approach the team has successfully optimised an enantioselective catalytic aldol coupling with isobutyraldehyde (figure 3). A chiral phosphine provides enantiocontrol, giving primarily the syn-aldol adduct in an excellent 87% yield and 99% enantiomeric excess.
Exploring various aliphatic aldehyde coupling partners typically delivers excellent yields and enantioselectivity, as long as a small amount (8mol%) of 2-methylthioethanol is added where required to maintain high syn:anti selectivity.
Surprisingly, when the team moves to aromatic aldehydes, the diastereoselectivity switches to favour the anti-aldol adduct, and the enantioselectivity is erased, delivering essentially racemic products. Introducing a much larger portion (30mol%) of 2-methylthioethanol reinstates some selectivity, with mechanistic studies indicating that the additive inhibits retro-aldol reactions and concurrent racemisation, likely by competing with the reaction product to bind the metal catalyst.
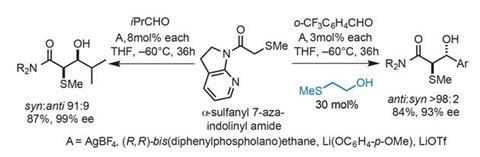
The scope of this reaction appears quite limited, especially as selectivity drops when moving away from the core substrates. That said, relatively few aldol acceptors appear in the paper, and it is unclear how far the team has explored. Furthermore, only two phosphine ligand variants are reported, suggesting the reactions could perhaps be further tuned.
Although the indolyl amide itself is not readily structurally modified, it behaves similarly to the synthetically useful Weinreb amides. It can be transformed into a ketone using organometallic nucleophiles, or reduced directly to an aldehyde or alcohol. The thioether group provides a handle for further derivatisation, and the team demonstrate a radical reduction and a radical allylation that proceeds with an intriguing retention of configuration.
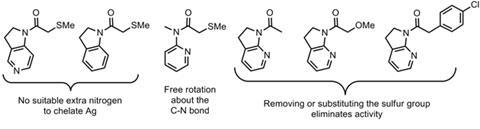
Applications for this system in complex molecule synthesis may not be particularly widespread. However, the idea of using an external additive to modulate the reactivity of the amide by restricting its conformation is much more interesting. Although direct evidence for this hypothesis is difficult to demonstrate, the fact that several related aldol donors all show no reactivity under the same conditions (Figure 4) suggests that it is definitely reasonable. The premise of externally induced conformational activation of unreactive C–H bonds is one that should be considered more widely.
Karl Collins (@karlDcollins) is a research associate at the University of Münster, Germany, and blogs at A retrosynthetic life











No comments yet