The 2015 chemistry Nobel prize was awarded to Tomas Lindahl, Paul Modrich and Aziz Sancar for DNA repair. Matthew Gunther reconstructs their story

The physical structures that surround us all exude a sense of stability – be it the homes we live in or the roads that we travel on. They are built with the best of intentions to stand the test of time, but are all vulnerable to the inevitable threats of aging and decay. To salvage them, we use the tools at our disposal to repair them, iron out flaws and keep them whole.
The 2015 chemistry Nobel laureates realised that the very molecular structure that makes us who we are, DNA, is locked in the same perpetual struggle. Tomas Lindahl, former director of Cancer Research UK’s Clare Hall Laboratories, Paul Modrich from Duke University in the US and Aziz Sancar from the University of North Carolina in the US were jointly awarded the prize ‘for their mechanistic studies of DNA repair’.

Their biochemical studies of life’s code have had a profound influence on the way in which we view not just the human genome’s vulnerability, but its capacity to fight as well. Back in the 1950s, this wasn’t an issue for the early pioneers of genetics. James Watson, Francis Crick and Maurice Wilkins, standing next to their newly discovered, double-helical model, were reassured this structure was stable, almost immutable, in the absence of external threats such as radiation. For Lindahl, this seemed puzzling.
‘In the Watson and Crick days, they discovered the double helix and that there’s this beautiful molecule that replicates faithfully – well, you know, it doesn’t,’ says Stephen West, Lindahl’s former colleague and now a group leader at the Francis Crick Institute, UK. ‘[Lindahl] realised that DNA was not as stable as people thought it would be – it’s not a lump of steel or like a brick, it is a substance that is very fragile.’
All about that base
Lindahl’s idea began to take shape when he was studying the conformation of RNA in the late 1960s. As a sideline to his research, he decided to isolate the polymeric molecule and heat it up to 90ºC. The RNA was irrevocably damaged after 30 minutes. Such a decay, he postulated, suggested the phosphodiester bonds that link nucleotide sugar units together, the very backbone of RNA, were being broken.
But it wasn’t initially a surprise to many chemists at the time. ‘Human beings have a lot of ribonuclease enzyme that breaks down RNA, so most people assumed I just had made a trivial observation on sloppy experimental work,’ Lindahl explains. ‘But then I did the experiment over a number of times, I made the RNA myself, so I was sure it wasn’t contaminated, and then I found the RNA slowly, slowly decomposed, always at the same rate.’ This observation was important, according to Lindahl, because it demonstrated there was a regular decay process occurring and wasn’t simply an artefact of contamination.
Following a short postdoctoral stint at Rockefeller University, US, Lindahl moved to the Karolinska Institutet in Sweden and was able to show DNA suffered a similar fate. ‘In Stockholm, I and my lab aid, Barbro Nyberg, who had fantastic lab technique, we could show DNA degraded spontaneously,’ recalls Lindahl.
Most people assumed I had made a trivial observation on sloppy experimental work
By labelling DNA in the gram-positive bacterium, Bacillus subtilis, with a 14 C radioactive tracer, Lindahl and Nyberg quickly established that purine bases, adenine and guanine, the molecular pairing and ordering of which gives us our genetic traits, leak from DNA. The release rate indicated that one molecule, normally vital for cell development, was responsible for the nucleic acids’ slow decay – water.
Within his bacteria samples, Lindahl noted that hydrolysis must be the culprit for this leakage. In every DNA nucleotide, of which there are billions in a single strand, there are three key parts: a nitrogenous base linked by a single glycosidic bond to a deoxyribose sugar and phosphate group. Lindahl discovered that this crucial glycosidic bond was being hydrolytically cleaved, leaving the affected bases to drift away from the safety of the double helix – a process known as depurination.
In addition to hydrolytic base removal, the spontaneous mutation of bases, be they cytosine or thymine, via the hydrolytic release of an amino group from a base was also uncovered by Lindahl. ‘One of the fundamental forms of DNA damage inside a cell is simply the decay of thymine bases,’ explains West. ‘So, the thymines tend to decay to form uracil – that’s an abnormal base in DNA.’
Cut, copy & paste
Such base anomalies are unable to hydrogen-bond with their previous base partner and resort to forming new bonds with adjacent bases. This results in a mutation that can be readily passed on through cell division and DNA replication. Lindahl estimated that up to 10,000 of these mutations, or lesions, occur in every healthy human cell every day. Left unchecked, such an assault on your DNA would mutate it beyond all recognition. The laureate soon discovered why we’re all still standing.1
Analysing Escherichia coli samples, he noted that the purine decay process he had first analysed in Bacillus subtilis was analogous to a mode of action in gram-negative bacteria by which a protein enzyme, N -glycosidase, was cutting deaminated cytosine from a nucleotide, leaving an empty base site.
Over the next two decades, and following a move to Cancer Research UK’s Clare Hall Laboratories as the site’s new director in 1986, Lindahl and his colleagues meticulously mapped out this repair mechanism, base excision repair (BER), reconstituting it in human cells.2
Like in E. coli, uracil–DNA glycosylase in human cells locates the uracil base by bending the DNA backbone for ease of access. The enyzme’s intercalation group flips the nucleotide base out of the helix before severing the glycosidic bond to remove the uracil. Another enzyme, endonuclease, then cuts the phosphodiester bonds around the remaining nucleotide to remove it before DNA polymerase is pasted into the empty site to produce a new base scaffold.
‘It’s the same principle as a dentist has when he fixes a hole,’ explains Lindahl. ‘He cleans up a little bit around the hole to get a clean area and then fills it in again.’
‘Your DNA is maintained in this very stable state because there is this kind of to-and-fro – there is the introduction of DNA damage and there is the recognition and removal of DNA damage,’ says West. ‘That’s what Tomas really realised before anyone else did.’
During this time, Lindahl, along with his colleague Richard Wood, now based at the University of Texas MD Anderson Cancer Center in the US, was also looking at other repair pathways, activated in response to external damage from UV radiation. ‘Tomas knew that I was interested in UV irradiation effects and he was particularly interested in nucleotide excision repair (NER), getting a system working to study the biochemistry of NER in human cells,’ explains Wood.
Swiss army knife
Lindahl and Wood’s subsequent research on NER, a similar mechanism in principle to BER, was greatly aided by the efforts of another 2015 Nobel laureate, Aziz Sancar, and his supervisor, Dean Rupp, at Yale University, US, in the late 1970s.
NER was already a known repair pathway by the time Sancar joined Rupp’s lab in 1977 after his PhD studies. 13 years earlier, Richard and Jane Setlow at Oak Ridge National Lab, US, had established that UV light can excite and break the hydrogen bonds between a thymine–cytosine base pair, giving rise to thymine dimers. These dimers introduce genetic mutations and physically deform the affected DNA strand. More important, however, was that the Setlows were able to show that these dimers are removed from DNA in E. coli, suggesting that an internal repair process was taking place.
NER works like a Swiss army knife
In the years following, Paul Howard-Flanders at Yale was able to isolate the three genes thought responsible for this, but no one was able to identify the protein repair enzymes encoded by them. If they could uncover the proteins at play, geneticists would be able to map out the entire NER pathway.?‘I felt, at that time, since very little progress had been made in elucidating NER and it had been known from the early days of Howard-Flanders’ genetic work, … it was my sense that cloning the genes would be a way to get at this,’ Rupp explains.
To achieve this, Rupp recruited Sancar, a promising researcher who had already cloned a gene responsible for UV-damaged DNA repair during his PhD. Through early discussions, the pair realised that if you can clone a gene within a cell and cordon it off from the rest of the main chromosomal DNA, it may survive and replicate after UV exposure. ‘This way we could label … the proteins made by the cloned genes and isolate them,’ explains Rupp.
Quite simply, it worked. Using [35 S]methionine, an amino acid tag, Sancar and Rupp were able to isolate the three repair enzymes involved in NER, the UvrA, UvrB and UvrC proteins, using this ‘maxicell’ method. The pair soon reproduced, in vitro, the main steps involved3 and found after further study that NER works like a Swiss army knife, with each protein having a different function and acting one after the other, to excise DNA damage.
For NER to be successful, an UvrA–UvrB trimer identifies the bump caused by a thymine dimer in the DNA. The UvrA leaves the damaged site, allowing the UvrC protein to bond with UvrB on the DNA strand. Both of these enzymes catalyse the breaking of phosphodiester bonds down- and upstream of the thymine dimer. The 12-base-long nucleotide is then removed from the site and polymerase again steps up to fill in the gap.
Sancar attempted to reconstruct this process in mammalian cells in the following years, with Rick Wood and his colleagues successfully doing so by 1995 at Clare Hall.
But for Rupp, perhaps their key contribution at Yale was in the cloning toolbox they created. ‘This was the most convenient way for identifying cloned proteins and, at that time, … there were really no other satisfactory, quick methods,’ he says. ‘There were certainly dozens, if not hundreds, of different gene products that used the maxicell method to identify the genes … not related to DNA repair at all.’
Genetic spellchecker
Although the maxicell method identifies specific proteins, in the 1980s Paul Modrich began to question whether DNA has its own machinery to identify bases and check whether they are in the correct location.
When DNA strands split and replicate during cell division, billions of bases have to be correctly aligned to ensure our genetic information doesn’t mutate and remains intact. But for every replication, hundreds of mistakes can occur whereby bases do not pair off with the correct partner.
Some mismatches are harmless to an extent and help to explain the diverse traits throughout the human race. ‘That’s one of the reasons you are you and I am me: we inherited from our parents very minor changes, most of which are not harmful,’ says Bob Lahue, a former colleague of Modrich, now based at the Centre for Chromosome Biology at the National University of Ireland Galway. He goes on to explain, however, that some mismatches can damage the genome and proliferate through DNA replication.
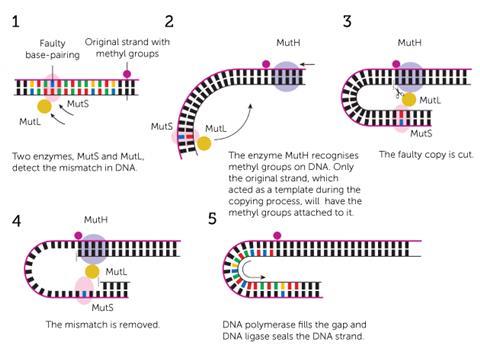
In the 1970s, geneticists had discovered that enzymes once again take up the call to repair these harmful genetic mismatches after being guided to a damage site by methylation signposts. Matthew Meselson, a geneticist at Harvard University in the US, and Modrich agreed that a synthesised DNA strand that is incorrect, as opposed to the complete original strand, may lack a number of methyl groups along the phosphate backbone. This would act as an indicator to a repair enzyme that could discriminate between methylated states.
At Duke University in 1989, Modrich and Lahue were able to identify the proteins responsible for following these methyl signposts and fully recreate the repair process in the test tube.4 ‘It’s a genetic spellchecker,’ explains Lahue. ‘So, just like when we type a document … it’s common at the end to go back and spellcheck [to] make sure you didn’t miss anything.’
This spellchecker consists of seven principal enzymes. A MutS protein latches onto the base spelling mistake and MutH binds at a methylated site further up the strand. MutL is the enzymatic bridge that links the two and directs MutH to cut the new, mismatched strand. This allows all of the enzymes to work together to unzip the two DNA strands and remove a large section of nucleotides containing the mismatch. Using the original strand as a moulding cast, polymerase forms a corrected nucleotide sequence in the empty space.
It’s a remarkably simple mechanism that bears a passing resemblance to BER and NER, and goes a long way to repairing those thousands of spelling errors after replication. ‘What mismatch repair does, this spellchecker, is go in after the fact and remove all but about 1 out of a 1000 [of those mistakes],’ says Lahue. ‘It reduces the error frequency by about 1000-fold.’
House of cards
But it isn’t foolproof. Knowledge of this repair system can help to highlight its flaws and how they can lead to hereditary diseases, according to West. ‘[Modrich] found this mismatch repair system in human cells and he also discovered that there are patients who develop bowel cancers,’ he says. ‘Patients with these bowel cancers are defective in this mismatch repair system.’
There are a couple of exceptions where mismatch repair is actually the bad guy
It’s a similar story for NER and patients who suffer from xeroderma pigmentosum, a disorder whereby the skin becomes extremely sensitive to UV damage and the sufferer can contract multiple skin cancers. ‘These patients are lacking one of the enzymes that catalyse this repair process that removes sunlight-induced DNA lesions,’ says West. ‘This [is] a direct relationship between a basic repair pathway and cancer.’
In some rare cases, the very machinery used to repair DNA can be turned against us, as researchers such as Lahue are only just beginning to discover. ‘There are a couple of exceptions where mismatch repair is actually the bad guy,’ warns Lahue. Geneticists believe that mismatch repair may actually help to induce Huntingdon’s disease, a neurodegenerative disorder. Certain DNA repair enzymes, such as Msh2, may latch onto a DNA strand in the wrong place and miss the mutated base pairs completely.
Such research may give the impression that the human genome is incredibly vulnerable, propped up like a house of cards. But, when called upon, the machinery Lindahl, Modrich and Sancar uncovered consistently patches up the cracks and stops the leaks thousands of times a day to preserve you – a stable house that DNA built.
References
1 T Lindahl, Proc. Natl. Acad. Sci. USA, 1974, 71, 3649 (DOI: 10.1073/pnas.71.9.3649)
2 Y Kubota et al., EMBO J., 1996, 15, 6662
3 A Sancar and W D Rupp,Cell, 1983, 33, 249 (DOI: 10.1016/0092-8674(83)90354-9)
4 R S Lahue, K G Au, P Modrich,Science, 1989, 245, 160 (DOI: 10.1126/science.2665076)












No comments yet