With the first approval of a drug containing deuterium looking imminent, Nina Notman surveys the deuterated drug landscape
Once inside the body, a drug molecule must dodge an onslaught of attacks by the metabolic system as it races towards its biological target. The body’s devotion to rapidly smashing drug molecules into tiny inactive pieces and then ejecting them means that many drugs need to be taken more frequently than they otherwise would.
One proposed way to tip the balance in favour of the drug is to substitute some of the hydrogen atoms in the molecule with heavier deuterium. Carbon–deuterium bonds are stronger than the equivalent bonds to hydrogen, leading to differences in reactivity that can be up to six to 10 times slower. So if breaking a C–H bond is responsible for a drug being metabolised faster than is ideal, substituting that hydrogen with deuterium may slow the molecule’s breakup. The end result being that the drug can be taken less often, say once a day rather than twice.
Because deuterium is an isotope of hydrogen this atom substitution causes no change in the shape, size, charge or the biological effects of the molecule. Only the pharmacokinetics, how the drug moves through the body, should change. And it’s not just drug half-life that can be improved by doing this: fewer drug–drug interactions, reduced variability in how the molecule metabolises between patients and improved tolerability can also potentially be achieved.
Deuterium is already widely used as an isotopic label to study the metabolism of drug candidates both in the lab and in humans. So far, however, it has never been used in a final drug molecule. But that might be about to change. Approval of the first-ever deuterated drug is now looking imminent. In August 2015, Teva Pharmaceutical Industries applied for permission to market a drug molecule containing six deuterium atoms in the US. The approval process typically takes around 10 months.
Slowing things down
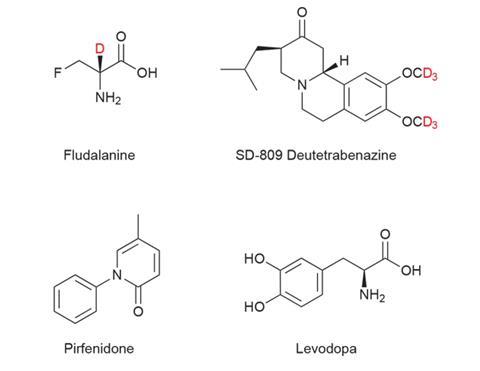
The idea of using deuterium to shield pharmaceuticals against unwanted metabolism has been bubbling away in the background for decades, and in the 1970s US pharma company Merck & Co put the first deuterated drug into clinical trials. The molecule fludalanine, or 3-fluoro-2-deutero-d-alanine, has antibacterial properties but problems with brain toxicity in Phase IIb trials meant work on the project ceased. The toxicity was unrelated to the deuterium.
There is a large variation in effect of deuteration, from really quite beneficial to nothing at all
About a decade ago, efforts to gain approval to market a deuterated drug started to ramp up again, when Auspex Pharmaceuticals (now owned by Teva), Concert Pharmaceuticals and Protia began applying for copious patents on deuterated analogues of known drugs.
‘It’s a great idea. You just change the pharmacokinetics, you know that the drug is going to work, and therefore you reduce the risk in the drug development process,’ explains Graham Timmins, a pharmaceutical scientist with an interest in isotope effects at the University of New Mexico, US. ‘The only risk is that your clinical effect is not going to be large enough to persuade somebody to pay a lot of money.’
Therein lies the rub: a C–H breakage is just one of many ways a drug can be metabolised, and figuring out a drug’s metabolic pathway (and therefore where to place the deuterium) is not always simple. ‘You’ll see this large range of variation of effect of deuteration from being really quite beneficial to doing nothing at all,’ says Timmins. A badly placed deuterium can even speed up drug breakdown.
Teva’s drug candidate currently undergoing review by the US FDA (Food and Drug Administration) is SD-809, also known as deuterated tetrabenazine or deutetrabenazine. Tetrabenazine has been used since the 1960s, when it was first licensed as an antipsychotic. Today, one of its approved uses is to treat chorea (involuntary movements) associated with Huntington’s disease. This is the first indication for which Teva is seeking approval to use its deuterated analogue.
Deutetrabenazine contains six deuterium atoms in two OCD3 groups on the aromatic ring. Teva report that the C–D bonds are eight times stronger than the C–H bonds in the parent molecule, and that the substitutions significantly increase the drug’s half-life in the body and offers other advantages including improved safety and tolerability.
‘Because we saw such a great improvement in the Phase I clinical trials between the deuterated compound and its proteo version, people couldn’t help but say, “Wow, that really makes a big difference”,’ says Thomas Gant of US pharmaceutical consulting firm Recondite Falls Discovery. Gant was the scientific founder of Auspex, but has since left the company.
Deutetrabenazine is also in trials against the movement disorders tar e dyskinesia and Tourette’s syndrome – while the undeuterated parent molecule tetrabenazine is not approved for either of these indications in the US, it is elsewhere in the world. The deuterated drug is in Phase III trials for tar e dyskinesia in the US, UK and EU. Teva say they hope to apply for FDA approval against this condition in 2016, but no details on plans outside the US have been announced.
When Teva purchased Auspex in May 2015, it said approximately 60 potential deuterated drug candidates had been identified by the company so far. Deuterated versions of pirfenidone (SD-560) and levodopa (SD-1077) being the other most advanced molecules.
Israel-headquartered Teva paid a massive $3.5 billion (£2.5 billion) to acquire Auspex, clearly signalling its belief that there is money to be made from souping-up known drugs using deuterium. Teva say they expect to have exclusivity of the acquired molecules in the US until at least 2031. Critically, all profit made is theirs alone: the owners of any still-valid patents on the undeuterated analogues of the molecules don’t get a look in – for the time being.
Improving compliance
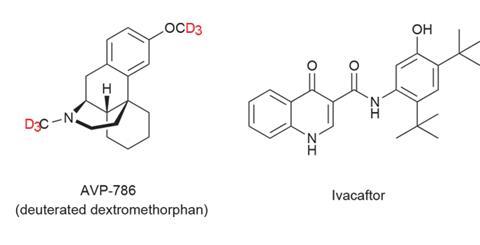
Concert, based in Lexington, US, is also attracting substantial investments and interest in its deuterated drug programme. The company’s most advanced drug is AVP-786, being developed in partnership with Avanir Pharmaceuticals (recently acquired by Japanese drugmaker Otsuka Pharmaceutical). AVP-786 is a deuterated analogue of the cough suppressant dextromethorphan, with both methyl groups fully deuterated.
Since 2011, dextromethorphan has also been approved in the US for use in combination with quinidine to treat the pseudobulbar effect, a condition often described as emotional incontinence, which can occur due to neurologic disease or brain injury. In the combination, quinidine acts to slow down the metabolism of the dextromethorphan. But quinidine’s side effects are of concern for patients with cardiovascular risk factors.
Avanir have now found that using AVP-786 reduces the amount of quinidine needed – as the deuterium is also working to slow down dextromethorphan’s metabolism. Less quinidine should mean fewer cardiovascular side effects. The AVP-786/quinidine combination is in Phase III clinical trials for treating agitation in patients with Alzheimer’s disease, and in Phase II trials in the US against major depressive disorder and schizophrenia.
This is a showcase for deuterium modification changing the pharmacokinetics of a compound
Concert is also developing deuterium-modified ivacaftor (marketed as Kalydeco by Vertex Pharmaceuticals). Ivacaftor is a new drug, first approved in the US in 2012 and then the UK in 2013 for treating cystic fibrosis. And this is the indication Concert is testing its deuterated analogue against.
Phase II trials are planned to start later this year. ‘We’re in the process of finishing up the Phase I programme with that compound,’ explains Roger Tung, Concert’s chief executive. ‘This is a really nice showcase for the ability of deuterium modification to change the pharmacokinetic property set of an existing compound.’
Ivacaftor is taken twice daily with fatty foods to help absorption. ‘The adherence to Kalydeco appears to be less than entirely optimal,’ says Tung. ‘We’re hoping that by simplifying the dosing regimen we’ll be able to increase the extent to which the in iduals with cystic fibrosis are able to adhere to the agent and obtain clinical benefit from it.’ There is no word on what Vertex thinks of this drug discovery programme because, if successful, it is fair to assume there will be a knock-on effect on ivacaftor profits.
Chiral control
Meanwhile, Andover, US-based DeuteRx is taking a different angle on deuterating drugs. Instead of tweaking drug pharmacokinetics, they are using deuterium to control a molecule’s chirality. ‘We are using deuterium to chiral switch racemic drugs that are developed as a mixture because the two enantiomers interconvert,’ explains Shelia DeWitt, DeuteRx chief executive. ‘By placing deuterium at the chiral centre we found that we’re able to slow down the racemisation and therefore have stereoselectivity, or stereo enrichment, of the desired enantiomers.’
Their signature approach is to place a deuterium at a chiral centre next to a carbonyl group to hinder formation of an enol intermediate. ‘Typically, in a basic environment a proton at the chiral centre alpha to the carbonyl will be abstracted. The planar intermediate, the enol, is then able to be quenched by water on either face which causes racemisation,’ she explains. As the C–D bond is stronger than its C–H equivalent, a hydrogen/deuterium substitution here slows down enol formation.
There’s a lot of precedent that a single enantiomer is always better than a mixture
DeWitt and her team have already proven this technique can make investors money. Her first deuterated drug company, Deuteria Pharmaceuticals, sold intellectual property rights for a compound to Celgene for a reported $42 million in 2012. It was the deuterated version of Celgene’s anticancer drug lenalidomide, a racemic mixture of S and R enantiomers that interconvert via an enol intermediate. Details of how the development of the deuterated analogue has progressed since purchase have not been announced.
‘There’s a lot of precedent that a single enantiomer is always better than a mixture,’ says DeWitt. ‘You always have a benefit in the pharmacokinetic profile any time you take a drug that’s a mixture of two compounds and select one of the enantiomers.’
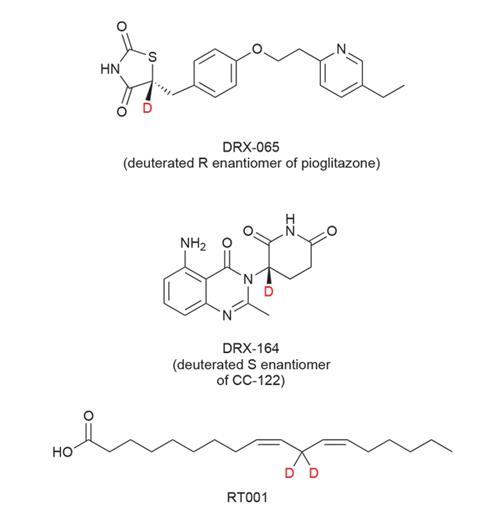
DeuteRx’s lead compound DRX-065 is the deuterated R enantiomer of the anti-diabetic drug pioglitazone, a racemic drug that has also demonstrated beneficial effects in a range of other indications including the rare inherited neurodegenerative disorder adrenoleukodystrophy (ALD). It is this condition that DeuteRx is pursuing for DRX065. Its preclinical work has shown that the S enantiomer of pioglitazone is responsible for the drug’s weight-gain side effect. ‘The R enantiomer exhibits the desired mitochondrial function modulation and anti-inflammatory activity without [the side effects]. We are planning to take DRX-065 into human clinical trials in Q3 of this year,’ says DeWitt.
DeuteRx are not just looking to tweak licensed drugs, they are also deuterating drug candidates still in the pipeline at other pharmaceutical companies. ‘DRX-164 is a deuterated S enantiomer of a drug candidate that Celgene has in clinical trials, known as CC-122,’ DeWitt explains. CC-122 is another thalidomide analogue and is tipped to become a more potent and safer version of lenalidomide. DRX-164 is still in pre-clinical research.
Patent worries
It’s not difficult to see why there have been bad feelings between some of these start-ups and the large pharmaceutical companies that funded the original drugs’ development. And things may get a lot more heated once the drugs start hitting the market place and pulling in big bucks.
‘Let’s say a big pharma company has got a patent on a non-deuterated version of a current non-generic drug. Then somebody else has got a patent that is issued on the deuterated version of that same drug. If the patent has expired, then there’s no point for big pharma to try and sue. But if they have got a valid patent, then that’s going to be interesting to see how that shakes out,’ says Timmins. ‘The deuterated drug won’t be 100% deuterated. It may be only 99% deuterated then that company will be selling 1% of something that’s covered by another person’s patent. Equally, the brand name company’s drug, there will be some naturally occurring deuterium in that. They will be selling that product that’s contaminated by a trace of the other person’s molecule.’
Big pharma are patenting the deuterated versions so other people can’t then claim them later on
To avoid this, many big pharma companies are starting to patent any potentially interesting deuterated analogues of all drugs in their pipelines. ‘Big pharma are claiming the deuterated versions so that other people can’t then claim them later on,’ says Timmins.
Pfizer is one such company looking to protect its assets in this way. The potential of deuteration is considered alongside most of Pfizer’s drug development programmes, explains Alfin Vaz, a drug metabolism chemist at Pfizer. ‘Most of our effort is directed at [seeing if deuterium can] change drug clearance times.’
The protection of Pfizer’s intellectual property may be the main purpose of this work, but development of a deuterated drug in the future hasn’t been ruled out. ‘All the ones that we have found where [deuteration] will help the compounds have not progressed for other reasons, not because of pharmacokinetics. Some day there may be a reason why we actually do bring forward a deuterated version of the drug, but at this point there is none in the pipeline.’
As well as big pharma better protecting their assets from the outset, Timmins expects that small companies will eventually find it harder to get US patents on deuterated analogues of established drugs because the patent office will start claiming the idea lacks novelty.
But Concert’s Tung says this isn’t happening yet: ‘We have been able to continue to get patents without much more difficulty now than we’d originally had overall. It’s pretty frequent that the initial response is rejection on the basis of novelty. Then we educate the examiner as to why this specific set of compounds that we are claiming have properties different from ones that others in that set share, and that these may not have been anticipated based on existing art.’
A different approach
One US deuterated drug company that could not be accused of lacking novelty nor stepping on someone’s toes is Retrotope. This Los Altos-based start-up is using deuterium to stabilise a membrane fatty acid against a natural disease-causing chemical reaction: oxidation. Lipid peroxidation, the oxidative degradation of lipids, has been cited as a probable cause of numerous medical conditions including Alzheimer’s and Parkinson’s diseases.
Linoleic acid, a polyunsaturated omega-6 fatty acid, is a primary component of cell membranes. This is an essential fatty acid that must be obtained from food because the body can’t synthesise it. Linoleic acid has two double bonds separated by a CH2 group. ‘That spot is normally a weakness because those double bonds want to be conjugated and they’re not. When a hydrogen at that spot is ripped off [by a reactive oxygen species], it becomes a radical – it is conjugated,’ explains Retrotope’s chief executive Robert Molinari. This radical then reacts with another fatty acid, and then many more fatty acids, until eventually it is quenched.
‘We focus on stopping the event that both initiates the entire cascade and causes it to propagate to other susceptible molecules in a dense packed membrane,’ he says. Retrotope’s lead drug is RT001, linoleic acid with two deuterium atoms on the carbon between the two double bonds. This molecule is taken orally and gets to the cell membranes via the natural fatty acid process.
In August 2015, the first clinical trial of this molecule started against Friedreich’s ataxia, a rare hereditary disease that causes progressive damage to the nervous system. ‘We have finished now the first cohort of low dose and we’re halfway through the high dose cohort at the moment,’ says Molinari.
In Friedreich’s ataxia, reduced expression of the iron-binding protein frataxin leads to free iron on the inner surface of mitochondria. And free iron is a catalyst of peroxidation of linoleic acid. ‘Having free iron on the inside of the mitochondria where the reactive oxygen species are being created is like having raw gas in a welding shop. It’s advancing the disease more rapidly,’ says Molinari. ‘While we do not change the amount of iron that’s in the mitochondria, what we can do is make the fat that starts this whole insidious auto-catalytic process fireproofed against radical attack.’
The decades long path to get a deuterated drug approved looks to be nearing the end, with Teva’s SD-809 seeming the most likely to get the first thumbs up. From an initial desire to extend drug clearance times, companies are now finding a broad range of potential uses for deuterium in drug molecules. In the future deuterium is expected to find itself embedded in an increasing number of the drugs that we see on the shelves of our local pharmacies.
Nina Notman is a science writer based in Salisbury, UK









1 Reader's comment