After some well-publicised problems, gene therapy – delivering DNA into people’s cells using viruses – is booming, Anthony King finds
Gene therapy is increasingly showing promise in clinical trials. Children with devastating diseases have had their lives transformed, yet the achievements are barely registering on the outside world, say researchers.
Gene therapy delivers a gene to a patient who either misses it or has a defective version, and the delivery vehicle (vector) is almost always a virus. The field suffered from hype in the 1980s and again in the 1990s, but is finally yielding results for patients, albeit in small numbers. ‘It’s almost like the public awareness has been inversely proportional to the success of gene therapy,’ says Simon Waddington at University College London, UK, who leads a team focusing on preclinical models for gene therapy. ‘Even scientists do not know how effective these therapies are becoming.’
A number of metabolic diseases caused by a faulty or missing single gene, often fatal or debilitating in early childhood, are the focus of clinical trials. ‘We’ve started to see a lot of industry involvement in gene therapy, which typically is a sign that things are going well. There’s a very different feeling in the field than there was five years ago,’ says Steven Gray, a gene therapist at the University of North Carolina in the US. What’s accelerated progress in the last five years is basic research into vectors. There are two distinctive strategies pursued, with two major types of viruses. ‘What we are seeing is payoff from a new generation of vectors,’ says geneticist Luigi Naldini, head of the San Raffale Telethon Institute for Gene Therapy in Milan, Italy.
Remove, fix, re-infuse
Lyososomes are sacs inside our cells with 43 enzymes, each of which dismantles certain molecules. Lysosomal storage diseases (LSDs) are caused by a mutation in a gene that leaves an essential enzyme dysfunctional; the target molecule builds up and causes problems in multiple organs and tissues.
We are gearing up for a clinical trial
Brian Bigger, University of Manchester
The diseases are considered ideal targets for gene therapy. Correcting the fault of one gene in some of a patient’s cells can be transformative, because a small subset of cells secreting the missing enzyme can correct the deficiency. Some LSD patients have received bone marrow transplants since the 1980s. In the traditional procedure, a chemotherapy drug kills bone marrow cells to make a space for stem cells from a donor without the faulty gene. These cells secrete the missing protein.
Gene therapy follows a similar tack, except a drug is used to coax the patient’s own stem cells into their blood, where they are filtered out. These are cultured and then infected with a retrovirus or lentivirus that has been rendered harmless. ‘The lentiviral vector integrates into the genome and expresses a transgene or missing gene with those cells,’ explains Brian Bigger at the University of Manchester in the UK. The patient’s altered stem cells are then injected back into them. His lab used lentiviruses to deliver a gene to mouse models of Sanfilippo disease (MPS-IIIa), a lysosomal storage disease. ‘We are gearing up for a clinical trial with a company we licensed the therapy to, Orchard Therapeutics,’ says Bigger. Progress in bone marrow transplants over recent decades has greatly assisted such therapies.
In Milan, the San Raffale Telethon Institute for Gene Therapy treats children with metachromatic leukodystrophy (MLD), a fatal disease involving a buildup of toxic lipids and which has no approved treatment. This devastating disease sees children lose speech and mobility by age two, then suffer from blindness, wasting and dementia; the children die around age five. The Milan results have impressed those in the field. ‘The treatment has been transformative,’ says Bigger. ‘They now have data for four years: all the patients attend school, have normal IQ and appear to have normal motor function, as long as they are treated before symptoms appear.’
Bursting the bubble
Remarkably, the stem cells put back into the patient after receiving the gene from lentiviruses cross the blood–brain barrier: normal enzyme levels have been measured in the patient’s cerebral spinal fluid. ‘If we get to the children early enough, we block the disease progression and the children we had treated are essentially healthy today,’ says Naldini, head of the Milan institute. ‘The advantage of gene therapy is that we use the patient’s own cells.’ Naldini’s institute found that up to 90% of the stem cells take up the gene from the vector.1 Key advantages include there being no risk of graft versus host disease or rejection of the transplant. Moreover, not all patients have a suitable donor for a transplant. Naldini also has used lentiviruses to treat patients with Wiskott–Aldrich syndrome, a severe combined immunodeficiency disease or SCID.2
In terms of our commitment to selling gene therapy, this is just the beginning
GlaxoSmithKline
SCIDs are known as ‘bubble baby diseases’, because children must sometimes be kept in sterile environments. In 1990, four-year-old Ashanthi DeSilva was the first patient to receive retroviral vectors; she received a transfusion of her own T-cells with healthy copies of the faulty genes in them. She survived. Not all early therapies worked out, with safety problems due to the retroviruses switching on cancer genes. Early versions integrated into the genome, flanked with gene promoters at both ends (known as long term repeats or LTRs). If the patient was unfortunate enough for this promoter to switch on a nearby oncogene, the patient could develop leukaemia. A recent survey by Bigger showed that 12 of 99 patients receiving these vectors got leukaemia, with two dying.
Newer viruses knock their own LTRs out and rely on a mammalian promoter. ‘Of these newer, self-inactivating vectors, 35 patients were treated and there have been no cases of insertional mutagenesis,’ says Bigger. The promoter can be chosen for the sort of expression needed, which could be moderate or high or tissue specific. Also, these ‘third generation’ lentiviral vectors prefer to integrate where there are genes rather than promoters. Most lentiviruses are derived from HIV-1, but only a small fraction of their genome is used to make the vector. ‘Starting from a deadly and fearsome human pathogen, they are extensively engineered to remove all gene and sequences not required for the virus to enter cells,’ says Naldini.
GlaxoSmithKline has licensed the therapy for MLD and it could be available next year. ’In terms of our commitment to selling gene therapy, this is just the beginning,’ says a GSK spokesperson. ‘We see this as a future platform that could stand beside vaccines, biopharmaceuticals, and small molecules.’ In the UK, Orchard Therapeutics is using a lentiviral vector to deliver the enzyme adenosine deaminase (ADA) to cells of ADA-SCID patients outside the body. There have been no problems of mutated genes in over 30 patients in the US and UK treated so far. ‘Monogenic metabolic disorders are the obvious place to start. We know the physiology of the deficiencies,’ explains Theodore Friedmann, a gene therapy pioneer at the University of California San Diego in the US. ‘One always learns an enormous amount studying rare orphan diseases.’
GSK sees their ex vivo work as a learning process. ‘The complexity of growing up a large batch of cells that can make the lentiviral vector is difficult because it wants to kill the cells you are growing it in,’ explains Jonathan Appleby, scientific advisor on gene therapy at GSK. Currently, all its gene therapy vectors are made in a facility adjoining the San Raffaele institute and patients must travel there to have their cells modified. ‘Vector manufacturing is challenging from a quality, time and cost perspective. We are working on ways to scale it up,’ says Appleby.
Into the body
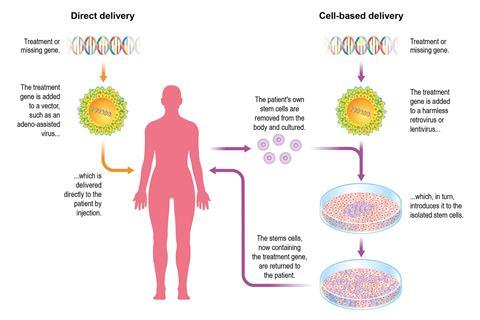
The second therapeutic strategy is to send in a viral vector to seek target cells and deliver a gene in the patient’s body. This might cure otherwise fatal diseases with a single injection. The in vivo strategy was marked, however, by a notorious and tragic case. In 1999, 19-year-old Jesse Gelsinger was treated with gene therapy for his rare liver disease in a US trial. He died from organ failure, probably as a result of an extreme inflammatory response to the adenovirus vector (common cold virus). Those in the field are familiar with the patient’s death and the mistakes leading up to it.
‘Every gene therapy trial was ordered to report their data and there was a flood of negative data, which had not been reported,’ recalls Bigger. The tragedy put the brakes on gene therapy trials in the US for 10 years. Some now see a silver lining in that pause, due to a refocus on patient safety. ‘A lot of investment went into really understanding the basic biology and the vector systems,’ says Gray. ‘The last five years we’ve seen a renaissance of gene therapy, with some quiet clinical successes adding to enthusiasm.’
Adenoviruses fell from favour. Instead, adeno-associated viruses (AAVs), which do not cause any disease in people, were tried. The AAVs deliver the therapeutic gene into a cell’s nucleus, but the circular DNA then sits outside the chromosomes. Because AAVs do not usually integrate into a cell’s DNA, this reduces the risk of mutations being caused in a patient’s DNA (known as insertional mutagenesis).
AAVs have led the way in fixing genes in vivo, starting with eye diseases. It is predicted that Spark Therapeutics will become the first gene therapy approved by the FDA for a gene therapy . Last year it reported that 27 out of 29 patients with inherited retinal disease had improved vision one year after treatment in a Phase III trial. ‘These children were destined to go blind but the treatment produced quite remarkable restoration of some functional vision,’ says Friedmann. Spark uses AAV vectors to deliver the gene to cells in the retina; the company is also starting clinical trials aimed at delivering to a gene to the liver of haemophilia A and B patients.
Targeting the brain
In 2015, Gray’s research led to a clinical trial for treating giant axonal neuropathy (GAN), delivering a gigaxonin gene using an AAV vector. GAN symptoms start in early childhood, initially with walking problems and worsen to include paralysis, seizures and dementia. The GAN research was funded by Hannah’s Hope Fund, set up by the Sames family in 2008 (their daughter Hannah has the condition). ‘We use AAV9 because it can cross the blood–brain barrier,’ says Gray. ‘We deliver it either intravenously or into the cerebral spinal fluid to concentrate it around the brain and spinal cord. It’s known that it targets neurons and astrocytes in the brain.’ AAV9 binds to receptors outside the blood–brain barrier and is shepherded across.
A search of people and non-human primates has turned up 100 AAV variants, now the workhorses of in vivo gene therapy. Some can target specific tissues, with AAV8 the vector for the liver and AAV9 for central nervous system disorders. In their spinal muscular dystrophy trial, biotech company Avexis uses AAV9. ‘Our recombinant AAV9 capsid delivers a functional copy of a human SMN [survival of motor neuron] gene across the blood–brain barrier to the patient’s own cells without modifying the existing DNA of the patient,’ says Brian Kaspar, chief scientific officer at Avexis. The downside of not integrating into the DNA is that the gene is lost if the cell divides – this makes it unsuitable for rapidly dividing cells like bone marrow, skin and gut cells. Lentiviruses are frequently the choice for such cells. However, neurons are extremely long lived, so the non-integration is not an issue when targeting the brain.
A one-off intravenous injection of AAV9 is also the strategy of Abeona Therapeutics in its gene therapy trial of Sanfilippo syndrome. While Lysogene in Europe uses a non-human primate AAV designated rh.10. ‘[AAVs] have become the most commonly used gene therapy vectors of for the central nervous system because of their safety, non-pathogenic nature and ability to infect dividing and quiescent cells in vivo, particularly neurons,’ explains Samantha Parker, Lysogene’s chief patient access officer. It administered the vectors directly into the brain of four patients, who all showed improvements in behavioural and sleep disorders. It plans a phase II trial but with a new promoter that Parker says increases enzyme expression around three-fold.
Improving potency and targeting
‘Over the last four or five years, there has been an exponential increase in the potency of these technologies,’ says Waddington. He moved the preclinical work forward using AAV vectors for haemophilia B towards the clinic. Between 2010 and 2012, Amit Nathwani at the UCL Cancer Institute and colleagues at St Jude Children’s Research Hospital in Tennessee, US successfully infused 10 men suffering from severe haemophilia with AAV8 vector to deliver a factor IX transgene, crucial in coagulation. This resulted in long-term expression at therapeutic levels of the missing factor, a first. Liver cells are long-lived protein factories that easily release their products into the bloodstream.
Waddington says such successes have been built on better vector targeting and efficiency. ‘I remember doing injections in mice around 2007 and seeing that they were expressing 3000% human factor IX,’ he recalls. ‘The perception at the time was that gene therapy was down, but we researchers in the field were motivated by the progress we saw.’
Crispr–Cas9 and gene editing in general can give us a higher level of precision when we correct diseases
Luigi Naldini, San Raffaele Institute, Milan
Different variants of AAVs go to different tissues and researchers tweak viruses to refine their targeting. ‘You can effectively target the brain, liver or muscles using different coat proteins,’ Bigger explains. Gray’s lab tweaks the capsid protein surrounding AAVs by trying different combinations. ‘One amino acid difference can change the effectiveness of an AAV,’ says Mark Kay, a gene transfer technologist at Stanford University in the US. ‘We and others are making libraries of capsid genes using multiple AAVs and then putting millions of variants under evolutionary selection.’ The best ones at targeting say liver cells are then sought by testing them on human liver cells put in a humanized mouse model. One chimeric capsid, made up of five different parent AAV capsids, proved dramatically more efficient at delivering a transgene into human liver cells than AAV8.3
Kay founded a company, Voyager Therapeutics, which aims to directly inject AAVs into the brain to treat patients with severe disease of the central nervous system. It has candidates at preclinical stages for Huntington’s disease, Alzheimer’s and monogenic amyotropic lateral sclerosis, a progressive and fatal neurodegenerative disease. Voyager’s lead candidate is for advanced Parkinson’s disease and a trial with five patients is under way. Other ways to improve the viral vectors include doubling up with two copies inside the AAV vector.
But there are still problems. Typically, 10 to 40% of us have specific immunity to AAV variants and produce antibodies to them. For now, patients who raise antibodies against a specific serotype are not treated with that viral vector, a problem when the viral vector cannot simply be changed (it’s part of the therapy and needs to be approved by regulators). ‘In future, we might be able to get around that through immuno-suppression or using toleration regimes of some flavour, but at the moment it’s a limitation,’ admits Bigger.
Sangamo Therapeutics in the US is using AAV viruses in clinical trials to deliver zinc finger nucleases to liver cells of patients with severe haemophilia B and so insert a gene precisely. It is also using these nucleases in clinical trials in patients with Hurler and Hunter syndrome, two lysosomal storage diseases. Zinc finger nucleases are looked upon by some as old hat, and it’s assumed viral vectors will eventually carry Crispr–Cas9 for gene editing. ‘The traditional approach of gene therapy using viruses to introduce functional genes will be with us for the foreseeable future, but gene editing is going to become a huge part of gene therapy, for treating and preventing a whole series of diseases,’ predicts Friedmann. ‘Gene editing means we don’t have to accept that the mutant gene stays in place. We can go into the cell and fixing the spelling of the relevant gene.’ Naldini agrees: ‘Crispr–Cas9 and gene editing in general can give us a higher level of precision when we correct diseases.’ GSK says it is adopting a ‘watch and wait’ approach to gene editing.
Last summer, 12-year-old Hannah Sames became the fifth child in a clinical trial to receive trillions of AAV viruses with the corrective gene for GAN. The trial continues, rooted in basic research on AAVs in Gray’s lab. Gene therapy is set to continue to deliver hope to rare disease patients, but it remains to be seen whether it can live up to its early promises and deliver cures to otherwise terminal or severely debilitating conditions. Gene therapy researchers are cautiously optimistic.
Anthony King is a science writer based in Dublin, Ireland
References
1 A Biffi et al, Science, 2013, 341, 1233158 (DOI: 10.1126/science.1233158)
2 A Aitui et al, Science, 2013, 341, 1233151 (DOI: 10.1126/science.1233151)
3 L Lisowski et al, Nature, 2014, 506, 382 (DOI: 10.1038/nature12875)












No comments yet