Andy Extance looks at two drugs that cranked the synthetic challenge all the way up to Halaven

Researchers have long sought to imitate the awesome organic synthesis power exhibited by nature in producing wildly complex molecules. Leading academics and their groups still slave to develop typically elegant, but usually still arduous, procedures for the glory of total synthesis. While chemists in the pharmaceutical industry might also be fascinated by these long routes, their costly and environmentally unfriendly nature generally makes them less commercially desirable than simpler compounds.
But sometimes fearsome molecules hold so much promise for use as drugs they can’t be ignored. For example, the anticancer potential of Taxol (paclitaxel) was first realised when it was extracted in the 1960s from the bark of the Pacific yew tree, Taxus brevifolia. But with six trees needing to be felled to get half a gram of the compound, testing would mean killing hundreds of trees.
While two groups first published different epic 40-step-plus total syntheses of taxol in February 1994,1,2 semisynthesis ultimately tamed the drug and allowed its commercialisation. A related compound, 10-deacetylbaccatin (10-DAB), can be isolated in high yield from needles of the European yew shrub Taxus baccata without destroying the plants. Robert Holton from Florida State University devised a four-step route from 10-DAB, including a reaction with a ß-lactam that itself takes several steps to prepare.3 In 1994, Taxol-maker Bristol-Myers Squibb commercialised this semisynthetic route to what would quickly become a blockbuster drug. In 2002, they switched to a plant cell fermentation process that produces paclitaxel directly.
Even without such shortcuts, some complex natural products are so useful they can support unwieldy production processes. Two such drug molecules, both discovered in ocean creatures, each require some of the most complicated chemical manufacturing today. This article celebrates the heroic synthesis efforts that brought them to market. Whether you want to improve the routes, or admire the doggedness of the scientists devising them, it’s hard to deny that they’re special.
Eribulin (Halaven)
Halichondrin B is a wicked molecule. In tests in mice, it is an extremely potent cancer cell killer, active at around 80 picomolar concentration. It also possesses a fiendish macrocyclic polyketide structure, with 32 stereocentres meaning that it could adopt over four billion different isomers – with just one that fights cancer.
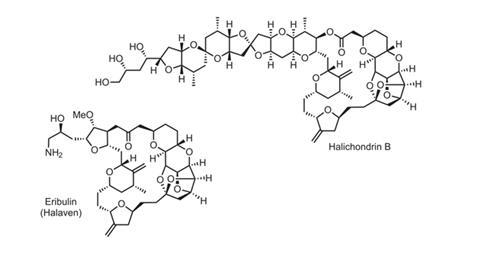
Its power is therefore inherently hard to harness. Halichondrin B was found in various sea sponge species in the 1980s, but getting 400mg of the compound from a tonne of sponge was doing well. Clinical development required at least 10g, and annual production takes kilograms.
Although developing a synthetic route to halichondrin B looked just as tough as trying to extract it from sponges, Yoshito Kishi’s group at Harvard University in the US accepted the challenge. Frank Fang, one of the team, recalls how the Nozaki–Hiyama–Kishi (NHK) coupling reaction would prove critical. ‘Another feature that was impressed upon me was the importance of crystalline intermediates,’ Fang adds. These allowed simple purification by recrystallisation, rather than expensive and time-consuming chromatography.
Published in 1992, their method used several NHK couplings, forming carbon–carbon bonds between multifunctional vinyl halides and aldehydes via a nickel-catalysed, chromium-mediated process.4 The sprawling convergent synthesis, whose longest linear sequence involved 47 steps, prompted Japanese pharmaceutical company Eisai to collaborate with Kishi in exploring halichondrin B’s structure–activity relationship. On screening the team’s intermediates, one featuring the macrocyclic half of halichondrin B proved especially active. A series of medicinal chemistry refinements led to what would eventually become eribulin (marketed by Eisai as Halaven), promising a slightly simpler synthesis. It has ‘just’ 19 stereocentres, which along with other structural restrictions cuts the possible number of isomers to a mere 16,384.
Fang joined Eisai in 1998 as it selected eribulin for further development, and worked to develop a production process for a route that produced it from three fragments. He again strove to exploit recrystallisation and use the NHK reaction, although making it reliable enough for manufacturing was challenging. ‘There was an appreciation for the somewhat sensitive nature of the reaction, particularly the asymmetric variant,’ he recalls.
The Eisai researchers therefore studied the NHK procedure as they applied it to redesigning the synthesis for part of the eribulin molecule they refer to as the C14–C26 fragment. Featuring just one ring, this fragment isn’t the most structurally complex of the three, but is still very difficult to make. That’s because it is a long chain with several stereocentres, whose stereochemistry is hard to link together.
Fang’s team initially broke this section down into two sub-fragments, C14–C19 and C20–C26, using asymmetric NHK reactions on each, learning about the reaction’s parameters as they did so.5 They then used what they’d found out to devise NHK reactions linking the two sub-fragments and attaching the two fragments on either side, which included closing the eribulin macrocycle. ‘We gained knowledge through our studies on the C19–C20 NHK coupling and were ultimately able to utilise that knowledge to try to execute an asymmetric NHK reaction in fixed equipment on multi-kilogram scale and construct the C19–C20, C26–C27, and C13–C14 bonds,’ Fang explains.
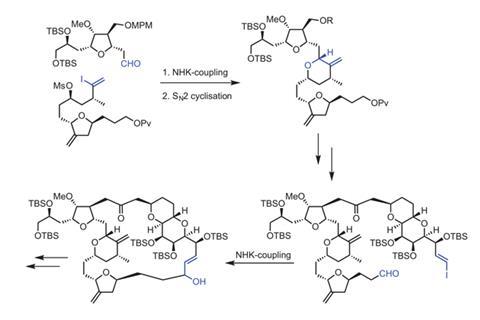
Halaven was approved in the US in 2010 to treat breast cancer and earned ¥2.89 billion in sales (£159 million) in 2014. The commercial route initially took 62 steps across a convergent synthesis bringing together three fragments, with a longest linear sequence of 30 steps. Fang’s team has since added seven steps to the C14–C26 fragment route, which counterintuitively cuts costs and waste by 80% by eliminating chromatography.6 ‘I am hopeful that we can find the lessons applicable in future work,’ Fang says.
Cheaper synthesis would appear welcome, given that Halaven’s price tag has been criticised. In the UK it currently costs £2,000 per 21 day treatment cycle according to data from the British National Formulary and the country’s National Institute for Health and Clinical Excellence (Nice). As a result, Nice refused to cover the drug, and in January 2015 the remaining funding in England looked set to be closed off with Halaven being taken off the Cancer Drugs Fund (CDF)’s list. But Eisai was told in March that the drug would stay on the list, pending reconsideration, after an appeal against the decision.
In defence, Fang claims that Halaven is actually one of the most affordable breast cancer treatments on the CDF. ‘Eisai was given no opportunity to lower the price of Halaven before NHS England announced that the treatment would be removed from the fund, despite this being something we were, and still are, very willing to do,’ he adds.
Trabectedin (Yondelis)
Though scientists first reported that Caribbean sea squirt extracts might grant cancer sufferers longer lifespans in 1969, it would be 38 years until that potential was realised. Sea squirts, gelatinous bag-like animals stuck to the sea floor, produce such tiny amounts of the compounds responsible that they couldn’t be isolated for over a decade. When these ecteinascidins eventually were identified, chemists found unique structures combining two or three tetrahydroisoquinoline rings and a 10-membered, sulfide bond-containing lactone ring.7
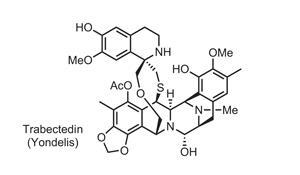
Kenneth Rinehart, of the University of Illinois at Urbana–Champaign in the US, who collected sea squirts while diving on reefs in the West Indies, patented the compounds and licensed them to Spanish company PharmaMar. To produce enough of the lead candidate, trabectedin, for clinical development, PharmaMar farmed and extracted over 250 tonnes’ worth of Ecteinascidia turbinata sea squirts. However, the complicated isolation and purification involved routinely reduced trabectedin yields below a gram per tonne of E. turbinata. Recognising the difficulty of this process, Rinehart asked E J Corey at Harvard University to develop a synthesis.
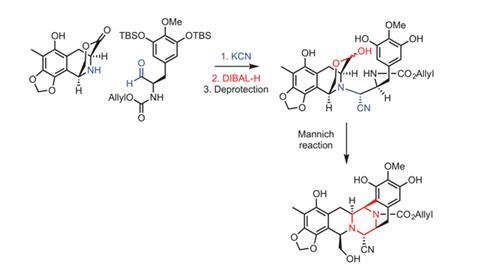
Corey’s team published a route in 1996 combining four fragments, involving over 40 steps, with an overall yield below 2%. One notable sequence forms a fused piperazine ring at the heart of the molecule, linking the first two fragments.8 The Harvard researchers exploited a Mannich reaction to weave together this complex junction, including joining two of trabectedin’s three tetrahydroisoquinolines. Later, to construct the 10-membered lactone bridge, they created a quinone methide (an exocyclic alkene conjugated into the ring) on one side of the appropriate tetrahydroisoquinoline. This could then be attacked by a sulfur atom at the end of a chain extending from the other side. ‘It was brilliant, but it would be impossible to have a product in the market without improving this chemistry,’ comments Carmen Cuevas, director of R&D at PharmaMar.
In 1999, amid PharmaMar’s efforts to develop an industrially useful process Cuevas started working on fermenting Pseudomonas fluorescens to produce the antibiotic safracin B. This molecule boasts much of the ecteinascidin fused ring structure, including the fused piperazine, rendering that part of the Harvard team’s inventive approach redundant. However, it lacks the 10-membered ring and is very unstable. Cuevas was able to resolve the instability, finding that introducing potassium cyanide at the end of the fermentation yielded the more robust cyanosafracin B molecule. ‘This reaction is around pH4, which means that you have hydrogen cyanide,’ Cuevas observes. ‘It was difficult to implement because there aren’t many people who want to work in these conditions.’
Under pressure to provide trabectedin for Phase III clinical trials, Cuevas was joined by two more PharmaMar scientists to adapt Corey’s procedure to start from this point.9 Yet the three would have to overcome a seemingly minor but surprisingly challenging structural difference – an amine in cyanosafracin B where the total synthesis called for an alcohol. In just a few months they revised the protecting group strategy, enabling the group to be converted, before working with a partner company to scale up the process. Although many of the steps, including forming cyanosafracin B, were hard to transfer from the lab, the method was industrialised before the end of the year 2001. ‘The speed of the process was a little crazy,’ Cuevas recalls. ‘I don’t know if we could do it again.’
Further tests would find that, whereas taxol and eribulin stop cells dividing by blocking their microtubules, trabectedin stops them multiplying by binding with their DNA. The two fused tetrahydroisoquinoline rings attach to a specific sequence in the minor groove of tumour cell DNA, causing it to twist abnormally. Normally, cell repair mechanisms would break the DNA chain at this point, but trabectedin’s third tetrahydroisoquinoline ring binds to the protein involved. The DNA damage is therefore permanent, triggering the death of the cancer cells.
Supported by a partnership with Johnson and Johnson that started in 2001, Yondelis first gained EU authorisation in 2007 to treat soft tissue sarcoma. It’s now sold in 80 countries, with worldwide annual revenues exceeding $100 million (£65 million). And while today some reactions have been improved to avoid chromatographic purification and isolating some intermediates, the route still involves 18 steps, Cuevas says, and is profitable. ‘The sales of Yondelis have put PharmaMar in the black after close to 30 years without anything in the market,’ she adds.
Andy Extance is a science writer based in Exeter, UK.
References
1 R A Holton et al, J. Am. Chem. Soc., 1994, 116, 1597 and 1599 (DOI: 10.1021/ja00083a066 and DOI: 10.1021/ja00083a067)
2 K C Nicolaou et al, Nature, 1994, 367, 630 (DOI: 10.1038/367630a0)
3 R A Holton, Method for preparation of taxol using ß-lactam, US Patent No 5175315, 1992
4 T D Aicher et al, J. Am. Chem. Soc., 1992, 114, 3162 (DOI: 10.1021/ja00034a086)
5 B C Austad et al, Synlett, 2013, 24, 0327 (DOI: 10.1055/s-0032-1317920)
6 F Belanger et al, Angew. Chem., Int. Ed., 2015, 54, 5108 (DOI: 10.1002/anie.201501143)
7 K L Rinehart et al, J. Org. Chem., 1990, 55, 4512 (DOI: 10.1021/jo00302a007)
8 E J Corey et al, J. Am. Chem. Soc., 1996, 118, 9202 (DOI: 10.1021/ja962480t)
9 C Cuevas and A Francesch, Nat. Prod. Rep., 2009, 26, 322 (DOI: 10.1039/b808331m)












No comments yet