Molecular machines have promised so much but are they more whimsical than technical? Philip Ball investigates
Molecular machines have promised so much but are they more whimsical than technical? Philip Ball investigates
Hardly an issue of any major chemistry journal passes without reporting some new molecular ‘machine’ or ‘device’. They are often staggering in their ingenuity and capabilities: they can crawl over surfaces, flex like muscles, even perform arithmetic and computation. They seem to tell us that chemistry has become a kind of scaled-down mechanical engineering.

But how far does that analogy really apply? And are these molecular machines anything more than just whimsical demonstrations of technical prowess? Indeed, are they truly machines at all? In a field that is arguably now at least two decades old, it may be time to take stock of what has been achieved, what the limitations are, and where the work is headed.
Moving parts
When surveying modern research on so-called molecular machines, the machine metaphor should not be confused with what really goes on at the molecular scale. When chemists talk of making molecular cars, elevators and trains they are offering a convenient picture of what they have devised; it doesn’t mean that they have exact, miniature analogues of these macroscale devices.
Take the molecular elevator devised by Fraser Stoddart, now at Northwestern University in Evanston, US, in collaboration with Vincenzo Balzani, Alberto Credi and colleagues at the University of Bologna in Italy. In this device, three hoops on the elevator stage are threaded by the tripod legs of the frame. Each hoop may be switched between two docking stages by protonation: by adding acid. It’s tempting to think that this means the elevator can be sent from one ‘floor’ to another on command. That’s kind of true, but it is really only an equilibrium ruffled by thermal fluctuations. At any moment, most of the elevators are at the bottom floor in acid and the top in basic conditions. But any specific elevator is liable to switch at any moment. Imagine that on the 33rd floor of the Hilton.
This points to the key distinction between molecular and macroscopic machines. Without fuel or power, the latter lie inert. But molecules are never still: under the influence of thermal noise and molecular collisions, they are constantly in motion, always changing shape. So molecular machines must, in effect, work in a maelstrom.
Clearly, this needn’t prevent things from getting done. In the cell, motor-borne cargo reaches its destination, valves and gates succeed in regulating trans-membrane traffic, life goes on. But it does so in a noisy manner. So what, then, can we expect of molecular machines, and what can we realistically do with them? And how can we design them to be reliable in the buzzing world of the molecule?
Shuttle service
The molecular elevator stems from one of the first molecular constructions to be presented as a mechanical device: the molecular shuttle, which Stoddart and his coworkers made in 1991 while he was at the University of Sheffield, UK. This was a rotaxane - a threaded hoop-and-axle molecule - with two potential docking ‘stations’ on the axle where the hoop could reside.
To turn molecular into large-scale motion, Stoddart and colleagues have coupled bistable rotaxanes to microscopic silicon cantilevers so that the switching makes the levers bend: a sort of molecular muscle. They strung two hoops on an axle with four stations, and chemical oxidation promoted movement of the hoops from the two end stations to the two in the middle. Because the hoops were covalently tethered to the cantilever surfaces, bringing them closer together tugged on the cantilevers, making them bend upwards by about 35nm - a much larger displacement than that of the hoops.
Jean-Pierre Sauvage at the University of Strasbourg in France has explored a related ‘molecular muscle’ concept in which two hoops are mutually threaded onto shafts attached to the hoop of the other molecule. The two hoops are pulled towards one another when the device is supplied with metal ions. This metal-triggered sliding mimics the way muscles contract by interdigitation of protein strands in the presence of calcium ions. Robert Grubbs and coworkers at the California Institute of Technology, US, have recently made ‘daisy-chain’ polymers in which a whole series of these units is linked together. In theory, Grubbs and colleagues estimate that a perfectly linear polymer of this sort should be 58 per cent longer in the extended state than when contracted. Over many monomer units, that difference will add up to an appreciable absolute displacement of the ends.
Open on command
Switchable molecular mechanics is itself old hat: heat and light, for example, have long been used to induce transitions between isomeric forms of a molecule that involve changes of shape. For example, a carbon-carbon or nitrogen-nitrogen double bond can be rotated 180° by ultraviolet (UV) light or heat, to give cis or trans isomers. In this way, the two groups are brought closer together or further apart. Jeffrey Brinker and coworkers at Sandia National Laboratories in Albuquerque, US, have used this principle to make light-switchable valves for opening and closing the channels of a porous form of silica. They coated the pore walls with azobenzene molecules, which have photoisomerisable nitrogen-nitrogen double bonds. In the cis form, produced by UV radiation, the pendant molecules are shorter and leave an open channel in the centre of the pores, whereas restoring the trans form using heat or green light switches the molecules back to a pore-blocking state.
While at the University of California, Los Angeles, US, Stoddart teamed up with Jeffrey Zink to devise similar switchable molecular valves based on pseudorotaxanes, in which the hoop can spontaneously unthread. They found that nanoscale pores could be blocked by a pseudorotaxane at the pore mouth, and opened by triggering release of the hoop. Stoddart and Zink recently made a more biocompatible version: hollow silica nanocapsules with porous walls through which the release of encapsulated molecules can be triggered by pH-dependent formation of threaded molecular complexes with hoops of sugar molecules called a -cyclodextrin ( a -CD). These sit on short stalks topped with aniline groups, attached at the pore mouths. When the a -CDs are threaded, the pores are blocked, preventing dye molecules inside from escaping. Adding acid protonates the aniline groups, which kicks off the a -CD caps, opens the pores, and releases the dye. This sort of controlled release might be used to deliver drugs to specific targets in the body, such as cancer cells.
Molecular ratchets

One thing that distinguishes these gates and valves from real machines is that they don’t get anywhere: the moving parts simply flip back and forth. In everyday engineering, we can turn that sort of repetitive movement into unidirectional motion: it’s what happens in a pendulum clock. The key there is the rack-and-pinion mechanism, which is a kind of ratchet in which asymmetric, sawtooth-shaped teeth only allow rotation in one direction. In other words, symmetric motion is made directional by an asymmetric geometry.
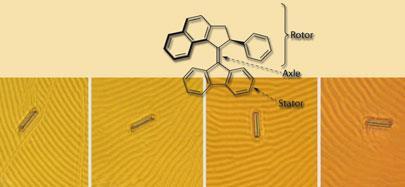
Ten years ago, Ben Feringa at Groningen University in the Netherlands realised that the rotation of molecular groups entailed by cis -trans isomerisation could be made directional by attaching an asymmetric structure. This enabled him to make the first unidirectional artificial molecular rotor from a molecule containing two identical propeller-like blades linked by a carbon-carbon double bond. The key was that the blades had to twist slightly to avoid bumping into each other at their ends, and this gave the whole molecule a twist: it was chiral. This biased the isomerisation process, so that continual irradiation with UV light could, if accompanied by enough heat, allow the molecule to rotate continuously in one direction. Feringa and his colleagues subsequently made rotors in which the two blades had different structures, allowing them to be targeted independently. They could fix one blade to a gold surface while the other blade was left to rotate freely. Fine-tuning this design has led to molecular rotors that will spin at nearly 10 million revolutions per second at room temperature.
Feringa and colleagues have used these motors to rotate much larger objects. They found that the light-triggered rotation could influence the molecular organisation of a liquid crystal film when the motors were added as a dilute dopant. This allowed them to rotate the fingerprint-like furrows that appear on the surface of the film. And a microscopic glass rod, 5 mm across and 28 mm long, floating on the top of the film, would ‘surf’ these undulations and rotate with them.
Feringa’s motors exemplify the principles for achieving unidirectional behaviour at the molecular scale: an energy source provides the impetus, and geometric asymmetry defines the direction. The classic example of this kind of process invokes a particle diffusing by random Brownian motion over parallel ridges. If the ridge profiles have an asymmetric, sawtooth form, then it is easier for the particle to move down the shallower slope than to surmount the steep one, and so there is net motion in one direction. This is called a Brownian ratchet.
The directional motion can’t be driven by thermal or Brownian fluctuations alone. Brownian ratchets need some external energy source such as a thermal gradient or a fluctuating fluid flow to drive the system away from equilibrium. At the molecular scale, chemical reactions can supply the necessary energy input, and this is how molecular motors and propulsion devices typically take advantage of the ratchet principle.
Brownian molecular ratchets are found in nature. The movement of RNA polymerase on DNA during replication seems to work this way, progressing steadily in the same direction.
Haiping Fang and colleagues at the Shanghai Institute of Applied Physics in China used Brownian ratcheting to propose a kind of molecular pump made from a carbon nanotube. They considered a nanotube embedded in a membrane matrix, through which water molecules can pass in a hydrogen bonded chain. Because water is polarised, three positive charges placed asymmetrically outside a carbon nanotube can pump water through the tube in one direction. These charges might be provided by chemical groups attached to the nanotube, or by tiny electrodes in the membrane matrix. Because the nanotube is so narrow, salt ions can’t pass through without losing their hydration shell. This costs too much energy, so salt is essentially excluded from the flow, making the construct (still hypothetical) a potential nanoscale desalinator.
It’s likely that motor proteins harness Brownian ratcheting to achieve directional motion. For example, the protein kinesin, which tows organelles as it ‘walks’ along microtubules, uses the binding of one of its two ‘legs’ to the track to bias the diffusional search of the other leg for a binding site in the forward direction.
Viola Vogel, now at ETH in Zurich, Switzerland, Henry Hess at the University of Washington in Seattle, US, and their coworkers, have shown that kinesin molecules anchored on a solid surface can pass microtubules around. They bound the kinesin to films of polytetrafluoroethylene containing striated grooves and ridges on which the proteins bind preferentially at low concentrations. This sets up linear tracks of motors for propelling adsorbed microtubules. The researchers later transported ‘cargo’ (nanoparticles) attached to microtubules.
The team has been putting its molecular transport system to ingenious uses. It exploited the random motions of fluorescently labelled microtubules to generate images of surface topography with a resolution of less than 50nm: because the microtubules can’t climb up sharp inclines, embossed patterns on the surface stay dark in time-averaged images. Hess and Vogel have grown microtubules from the component tubulin proteins within microfabricated polymer channels on glass, thereby creating patterned tracks on which they use kinesin to transport fluorescent nanoparticles in a directional manner. In this way they made molecular roundabouts which look like highway roundabouts seen from above at night.
And last year Hess and his coworkers described a ‘smart dust’ sensor device in which microtubules would capture an analyte in one region and carry it over a kinesin-coated surface to another region for fluorescent labelling, and then to another for detection with light. This removes the need for separate washing, tagging and detection steps in conventional assays: the molecular machinery does it all.
DNA on the move
So if we are interested in getting the job done rather than in exploring and extending the limits of synthetic chemistry, it might make a lot of sense to use nature’s molecular machines. ‘Motors are involved in nearly every key process in the cell and living organism,’ says Feringa, for example in cell division, motion, signal transduction and mass transport. And they are efficient devices. ‘I can do things with kinesin motors which are orders of magnitude faster, more specific and more powerful than what one can do with rotaxanes,’ says Hess. However, he admits that ‘my hybrid solutions are still far away from application-ready, due to their limited stability’.
DNA offers perhaps the best of both worlds. It can be assembled, manipulated and even replicated with existing biological components. And it can be twisted and bent and looped into all manner of shapes. It has already been used to make a wide variety of molecular machines. For example, Bernard Yurke of Bell Laboratories in Murray Hill, New Jersey, US, Andrew Turberfield of the University of Oxford, UK, and their coworkers, made a DNA machine shaped like a pair of tweezers that could be switched between a compact and open form by adding two single DNA strands in succession. A ‘fuel’ strand binds to part of the machine to which its sequence is complementary, producing a shape change. It is then peeled off to restore the initial conformation by the second strand, a kind of ‘anti-fuel’. The fuel strand has a few bases left dangling on which the anti-fuel strand can get a foothold to start stripping it away.
The researchers subsequently figured out how to make DNA machines that switched their conformation repeatedly in a free-running manner when provided with the fuel and anti-fuel strands, without the need for the experimenter to intervene constantly to complete the cycle.
Nadrian Seeman at New York University, US, and his colleague William Sherman have used the same principles to make a DNA biped - two double helical legs connected by flexible linkers and ending in single-stranded feet - that ‘walks’ along another DNA strand. The footpath contains a series of toeholds with dangling single strands, to which one of the biped’s feet can be bound by adding a set strand on which half the sequence is complementary to the free strand on a particular toehold and the other half is complementary to the dangling foot sequence. The foot is released by adding an unset strand that peels off the set strand, starting from a region left unpaired at the end of the foot.
If, however, the toeholds of the path are all equivalent, then a walker of this sort will simply move at random along it. And if the detachment of one leg is not coordinated with the attachment of the other, there’s no guarantee that the walker will remain fixed to the path at all. These limitations have recently been overcome by Seeman and colleagues, who designed a walker in which the motion of the legs is coordinated: the leading leg catalyses the release of the trailing leg. This stepping cycle is triggered by fuel strands, and it relies on the track itself being asymmetric, with non-equivalent toeholds. In this way the DNA walker becomes a Brownian ratchet. It’s an ‘expensive’ stroll, however, because each toehold is in effect destroyed when stepped on: the walker burns its bridges. So we have a way to go yet before we can mimic the coordinated motion exhibited by kinesin on conserved microtubule tracks.
Working together
Feringa foresees a shift in chemistry and microphysics, away from static, equilibrium systems and towards dynamic ones. ‘Out-of-equilibrium, kinetically driven, dynamic systems governed by switches, triggers, response elements and motors will change drastically our approach to smart, responsive and adaptive materials, devices and sensors, computation, and so on.’
Now that molecular engineers are almost spoilt for choice, they need to consider their options carefully. What are the most robust and reliable fabrics for making these devices, and how might they best be powered? Balzani thinks that light-driven systems have several advantages. ‘All living organisms rely on sunlight as the primary energy source,’ he points out. Light is clean and abundant. In contrast, ‘a device that utilises chemical energy will need addition of fresh reactants at any step of its working cycle, with the concomitant formation of waste products.’ What’s more, he says, the energy input can be carefully controlled by the wavelength and intensity of the exciting photons. And this energy ‘can be transmitted to molecules without physically connecting them to the source - no ’“wiring’’ is necessary’.
Stoddart thinks that the most useful machines may be ones that, while perhaps inspired by nature, do not follow their design too slavishly. ‘My chemist’s view is that we need to go down a road involving construction that is much more robust in a skeletal sense than we see in nature,’ he says.

And how might we want to use such machines? In biology, they may power macroscopic devices, such as an arm or leg. But for applications like that, three key questions arise: how much force can they produce, and how quickly, and at what energy cost? Initial rough estimates don’t bode well. For example, a polymer molecule containing 20 light-switchable azobenzene pivots can perform only about 4.5 x 10-20J of work per azobenzene, which is scarcely more than the thermal noise. However, other devices pack more of a punch: a switchable rotaxane might produce a factor of several tens of the thermal energy per molecule. And if large arrays of molecules can be organised so that they all act in concert, as they do in muscle, then the collective force can be substantial.
Considerations like this lead Stoddart to think that molecular machines whose purpose is to induce motion at scales much larger than themselves will have to be organised into large assemblies. ‘Single artificial motors swimming around aimlessly in solution are going nowhere fast,’ he says. ‘We have really got to move on into new arenas, into extended structures, into one- and multi-dimensional polymers, onto surfaces, into interfaces, and so on. Intuitively, one feels as the cooperative systems grow in size, the thermal fluctuations that undoubtedly dominate the small molecular world will surely start to become less important, and ultimately will more or less peter out.’ Then we’ll be getting somewhere.
Further Reading
J D Badjic et al, Science, 2004, 303, 1845
S Silvi, M Venturi and A Credi, J. Mater. Chem., 2009, 19, 2279
P G Clark, M W Day and R H Grubbs, J. Am. Chem. Soc., 2009, DOI: 10.1021/ja905924u
R Hernandez et al, J. Am. Chem. Soc., 2004, 126, 3370
M Klok et al, J. Am. Chem. Soc., 2008, 130, 10484
H Hess et al , Nano Lett., 2002, 2, 113
V Balzani, Chem. Soc. Rev ., 2009, 38, 1542










No comments yet