Simple method for identifying incorrect structures of organic molecules due to NMR misassignments
Imagine finally completing a 30 step total synthesis only to discover that the molecule you were aiming for was the wrong one. The consequences of structural misassignment of complex organic molecules can be costly and time consuming, not to mention frustrating. Now, a new NMR method aims to highlight errors in proposed structures at a much earlier stage, preventing such scenarios.
NMR spectroscopy is a standard tool for elucidating the structure of organic molecules. This may be a straightforward job when confirming the identity of small molecules. However, in the case of complex molecules, the task becomes much more difficult and errors can result in the wrong structure being proposed.
Ariel Sarotti from the Rosario National University, Argentina, has developed a new, computationally inexpensive method combining calculated and experimental 13C NMR data to flag up incorrect structures. This rapid and simple process can determine if a candidate structure is incorrect, using trained artificial neural networks (ANNs) to find patterns in both the calculated and experimental data to do the decision making. A set of 200 molecules with known correct and incorrect NMR assignments was used to create and train the system. The subsequent testing phase correctly identified the incorrect structures of a set of 26 natural products. While some knowledge of computational chemistry is required, Sarotti’s development of an Excel spreadsheet tool will allow chemists to use the method without being experts in ANNs, making it much more accessible.
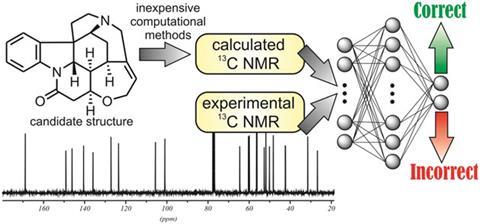
Current methods rely on comparing multiple sets of data for at least two possible structures. ‘These approaches crumble when only one structure is available, as there is nothing to compare,’ says Sarotti. Significantly, his method uses only a single set each of experimental and calculated data. As Jonathan Goodman, whose research at the University of Cambridge, UK, focuses on investigating organic structure and reactivity, using experimental and computational methods, explains ‘if you put less information in, you get less information out, so this is a much harder question, but that’s what this paper does and it seems to work rather well.’
Sarotti mentions that his method has limitations, as it does not necessarily confirm that the proposed structure is correct. Instead, it is designed to identify when there might be something wrong. This point is particularly relevant when dealing with cases of stereoisomerism, one of the most challenging aspects of structural assignment and one that Sarotti hopes to tackle in the future.








No comments yet