
Researchers have developed a new reaction that converts chiral molecules into the opposite stereoisomer. It works on unactivated stereocentres that until now have been difficult to target. The method could allow researchers to access new molecular configurations and could alter the way in which chemists plan their total syntheses.
When chemists set out to build complex chiral molecules, they tend to use sequences of reactions that are designed around constructing chiral centres. This is often the most challenging aspect of building up complexity during a synthesis project, notes Alison Wendlandt from the Massachusetts Institute of Technology, US.
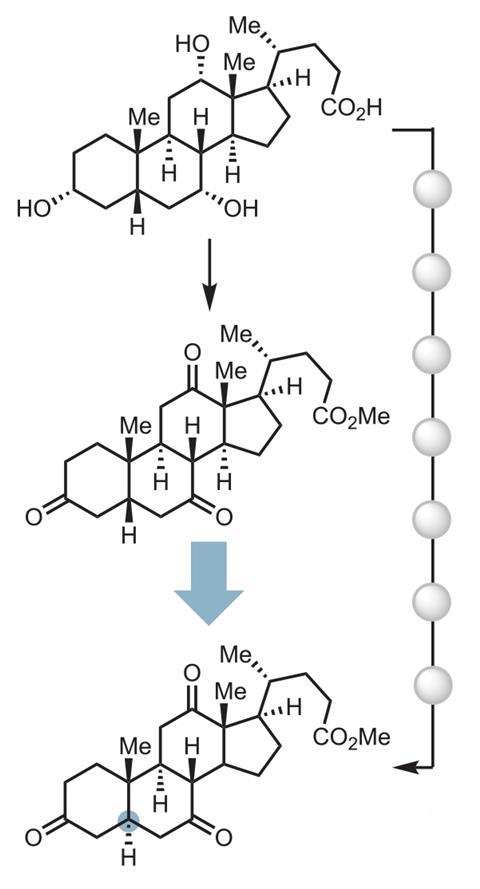
Now, Wendlandt’s team has found a way to build diastereomers of complex, drug-like molecules in a strategically different way. ‘Our tool essentially allows us to try to tune the three-dimensional structure in a late-stage setting,’ she explains. ‘And so [if] you have one diastereomer, you can now access a completely different diastereomer without having to go back resynthesise the molecule with the correct stereoform that you wanted.’ This means when planning a synthesis, chemists can now decouple bond-forming or complexity generating steps from the steps that are needed to gain three-dimensional control, says Wendlandt.
The technique relies on a polyanionic tungsten photocatalyst and disulfide co-catalysts that promote epimerisation reactions, as in those that invert the configuration of a chiral centre. Wendlandt’s team previously altered stereochemistry in sugars, and has now shown that it works on a much broader range of molecules containing unactivated tertiary carbon stereocentres.
‘Where I think this will really shine is in the medicinal chemistry application,’ says Wendlandt. ‘It’s really hard to predict what the effect of certain stereocenter will have again on the interaction of that small molecule with, say, an enzyme or a protein – and so, quite frankly, we just have to make all of the diastereomers of a chiral molecule and see what happens.’
Using this stereoediting logic, drug discovery chemists could simply synthesise the diastereomer that is easiest to make and then quickly access the opposite isomer using Wendlandt’s photocatalyst. ‘Even if you get a 10% or 20% yield of a target, that’s enough to validate: Is that a better drug candidate? Or is it a worse drug candidate? And that can save you a tonne of time,’ says Wendlandt.
University of Wisconsin–Madison, US, photocatalysis expert Tehshik Yoon describes the findings as ‘remarkable’. He notes that while numerous strategies exist to epimerise stereocentres that are next to polar functional groups, ‘the ability to alter stereochemistry at an unfunctionalised carbon in a complex molecule is extraordinary’.
‘I think it could impact synthetic chemistry in a lot of ways, but I’m most excited by the way in which it diverts methods that produce well-established stereochemical relationships, [for example] the Diels–Alder reaction, towards stereochemical outcomes that would otherwise be really challenging to produce,’ adds Yoon. ‘That the method uses common, well-accepted reagents just makes the innovation even more impressive and probably easier for the rest of the field to adopt.’
References
Y-A Zhang et al, Science, 2022, 378, 383 (DOI: 10.1126/science.add6852)












No comments yet