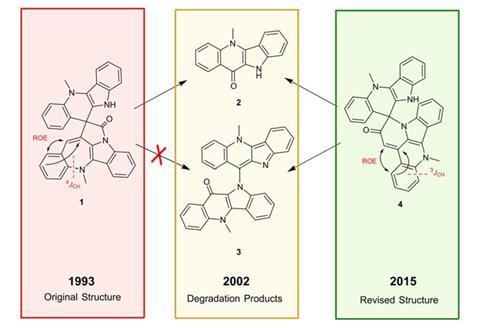
An adapted protein NMR technique could spell the end for complex molecular structures that are later shown to be incorrect
Nuclear Magnetic Resonance (NMR) spectroscopy is perhaps the most useful technique for determining the structures of molecules that do not form suitable crystals for x-ray crystallography. However, it has been known to produce incorrect structures, particularly for complex molecules. But now researchers at Merck & Co., along with academic partners in the US and the Netherlands, have adapted a method more commonly used in biological studies so that it can be used to check whether a proposed structure is correct, or pick the right candidate from several possible structures.
NMR works by placing molecules in a magnetic field, and measuring their radio-frequency absorption spectrum – the frequency required to flip each 1H nucleus, for example, from the parallel (aligned with the field) to the anti-parallel state. This can be used to infer the chemical environment of each nucleus and solve the molecular structure. In practice, however, the structures of large molecules are tricky to resolve accurately because different atoms may be in similar chemical environments and will therefore have a similar chemical shift, making unambiguous assignment difficult or impossible.
But a team led by Yizhou Liu of Merck’s Structural Elucidation Group may have suggested a way around this. They showed that incorporating the molecules of interest into a polymer gel that has been constrained – either stretched or compressed – can open up two further sources of information, as the molecules within the gel’s pores align along the constraining direction. The first is the residual dipolar couplings (RDCs): typically the couplings between 1H nuclei and those of carbon atoms bonded to them. These vary in a calculable way with the angles between the carbon-hydrogen bonds and the applied magnetic field. The second is the residual chemical shift anisotropy (RCSA): ‘In general, electrons are not evenly distributed around the nucleus due to the defined geometry of chemical bonding – the chemical shift is field-orientation dependent’ explains Liu. ‘In liquids, the chemical shift is averaged to an isotropic value due to fast random molecular rotation. When the molecule is partially aligned in a gel, some orientations are more likely than others, and the average chemical shift moves toward values corresponding to the preferred direction – therefore you have a change in observed chemical shift.’
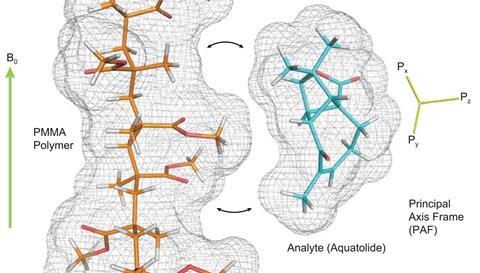
Both the RDCs and the RCSAs can be calculated for each candidate structure and compared to the experimental results. ‘If you plot the calculated versus the experimental data and you’ve got the correct structure,’ explains Gary Martin, another member of the Merck team, ‘then all of the RDCs and RCSAs are going to be consistent with the experimental data and you’ll get a relatively straight line. If your data is more scattered, then some feature of your structure is incorrect or problematic.’
The researchers looked at several molecules – cryptospirolepine, aquatolide and products of the rearrangement of a spiroketal molecule – whose structures have all been revised recently. In each case, they used a combination of density functional theory and computer-assisted structural elucidation software to calculate theoretical RDC and RCSA values for various proposed structures. They then conducted experimental tests on each molecule and found that, by comparing theoretical and experimental values, they could identify the correct structure. Martin notes that the structure of aquatolide was originally identified incorrectly and required years to solve, using computational analysis of over 60 different structural possibilities, x-ray crystallography and finally total synthesis. ‘Using RCSA and RDC data you can get to the same place in about a week given a couple of milligrams or even a milligram of the compound,’ he says.
Ad Bax of the National Institute of Diabetes and Digestive & Kidney Diseases in Maryland, US, says that although the techniques showcased here are in wide use in protein NMR, the paper will probably be ‘an eye-opener for the regular organic chemistry community’. ’They’re addressing a different problem really. They’re asking “What is the molecule?” [In protein NMR] we already know what the molecule is typically from the gene sequence,’ he explains.









No comments yet