
Two researchers in Spain have analysed the occupation of carbon atoms’ effective atomic orbitals to create a framework for quantifying substituent effects in aromatic systems.1 By linking electronic structure to inductive and resonance effects, the framework could enable chemists to better understand and predict Hammett parameters.
Since its development in the 1930s, the Hammett equation has been a powerful tool for rationalising and predicting the reactivity of organic molecules, with Hammett parameters indicating the electron-donating or electron-withdrawing nature of aromatic substituents. ‘Hammett relationships have been very, very important in the development of organic chemistry … what has been rather difficult is to rationalise them on purely theoretical grounds,’ comments theoretical chemist Ángel Martín Pendás from the University of Oviedo in Spain, who wasn’t involved in the work.
Now, Pedro Salvador and Gerard Comas-Vilà at the University of Girona in Spain have used effective atomic orbital2 analysis to produce charge-density-based descriptors, IX and RX, to respectively quantify inductive and resonance effects exerted by substituents in aromatic systems. ‘What we did was single out, from the total density of the molecule, which part you associate to each of the atoms, and then made orbitals out of it,’ explains Salvador. ‘The rationale is that when you have a substituent, the shapes of these orbitals remain the same essentially, but the occupations change.’
Computational studies of a chemically diverse set of mono-substituted benzene derivatives demonstrated that the inductive effect of a substituent could be accurately represented by changes in the occupation of the effective atomic orbitals involved in the C–C bonding framework at the meta position. The resonance effect, meanwhile, is well reflected by changes in the occupation of 2pz-type effective atomic orbitals on carbon atoms located at the ortho and para positions.
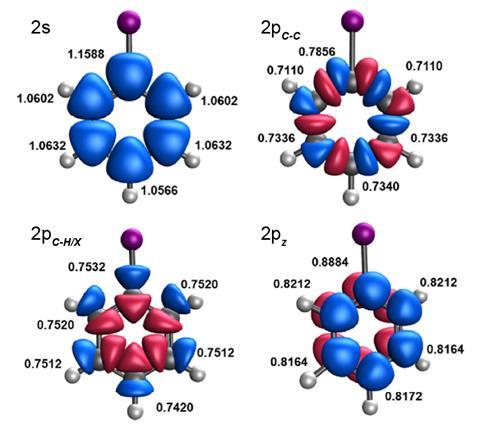
Salvador and Comas-Vilà used their descriptors to predict Hammett parameters for meta- and para-substituted benzoic acid derivatives. The resulting values correlated extremely well with previously reported values – for meta-substituted derivatives the mean average error was just 0.04. ‘The most exciting part for me was to compare our results with experimental results, and see that they fit really well, the correlation is excellent,’ says Comas-Vilà.
This new framework can serve as a tool for predicting Hammett parameters in aromatic systems, whose experimental determination can be time-consuming and resource-intensive. It also provides a more comprehensive understanding of electronic interactions in substituted aromatic systems, aligning with a central goal of Salvador’s team: connecting classical chemistry concepts with advanced quantum mechanical techniques. Martín Pendás also appreciates this aspect of the work, noting that ‘Hammett parameters can be sensed by the change in the number of electrons that you find in each of these orbitals… I think that they correlate fantastically well with the parameters, so they now have a method to find at which point in the molecule you see an effect from changing a substituent for another substituent.’
References
1 G Comas-Vilà and P Salvador, Phys. Chem. Chem. Phys., 2025, DOI: 10.1039/d5cp01299f
2 I Mayer, Int. J. Quantum Chem., 2014, 114, 1041 (DOI: 10.1002/qua.24623)




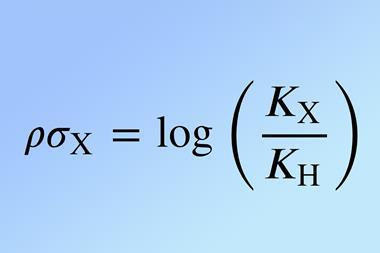








No comments yet