Technique can slow metabolism of medicines, giving them more time to work
US chemists have found a neat and general way to add ‘unnatural’, strained ring systems to drug molecules that could help boost efficacy by slowing clearance from the body.
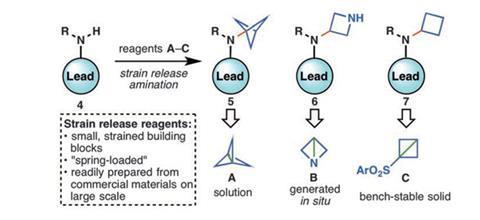
Medicinal chemists have known for some time that introducing a small strained ring into a drug skeleton can make them less recognisable to our metabolic enzymes. As a result metabolism of the drug is slowed giving more time for the physiologically active part of the structure to do its job.
Unfortunately, incorporating bicyclo[1.1.1]pentanes, azetidines or cyclobutanes into a drug molecule is often tougher in terms of the overall synthetic scheme than building the basic skeleton of the drug itself. Indeed, this obstacle has led to promising drug leads being abandoned because of synthetic difficulties.
Now, Phil Baran of the Scripps Research Institute in La Jolla, California, and colleagues there and at Pfizer have developed a new approach to appending such strained groups on to core scaffolds by ‘spring loading’ carbon–carbon and carbon–nitrogen bonds in the skeleton. Although such efforts have been attempted before, their use of amine nucleophiles and the spring-loading approach precludes the need for more than a single additional step in the synthesis, thus keeping costs and complexity down and making the process amenable to pharmaceutical production. Even optimised early efforts required three steps.
The team explains that their strain-release amination involves a process-friendly amine synthesis and side-steps the usual intermediates on which an organic chemist might alight through retrosynthetic analysis of the target. The approach exploits the most strained bond in propellane and any of a selection of strain-release agents, the use of which can generate the amine derivatives with the small unnatural cyclic group and so open up the drug discovery space enormously.
‘This work lays a foundation for the synthesis of new chemical entities, probe molecules and click-like connections that rely on the native activation of strained C–C bonds,’ the team writes. They also point out that their strain-release agents are easy to use. They also require only mild reaction conditions, are inexpensive to prepare and are chemoselective and can be used not only for drug lead optimisation but also peptide labelling and bioconjugation. ‘We will further extend the range of chemistry that can be accomplished with these strained systems,’ Baran tells Chemistry World.
Ganesan of the school of pharmacy at the University of East Anglia, UK, explains how aromatic or heteroaromatic compounds are popular in drug discovery but the resulting flat molecules are often correlated with what he refers to as ‘pharmacokinetic liabilities’. ‘Baran’s elegant methodology facilitates the introduction of small ring non-flat sp3 hybridised building blocks into synthetic schemes,’ Ganesan says. ‘The availability of these building blocks and their ease of synthesis will encourage medicinal chemists to explore them in a variety of applications.’
References
R Gianatassio et al, Science, 2016, DOI: 10.1126/science.aad6252











No comments yet