Meet the organic chemists intent on breaking bonds that are generally considered to be unreactive
The established logic of organic synthesis revolves around functional groups, where reactive groups of atoms such as carbonyls, alcohols and amines control which bonds in a molecule will break under certain conditions. The art of synthesising a complicated molecule is being able to orchestrate these in the most effective way during the process of building up a molecule from a simple starting material.
Carbon–hydrogen bonds are deemed unreactive – unless activated by being near a functional group – and therefore historically ignored when planning a synthetic route. But in recent years there has been a paradigm shift in the logic of synthesis. ‘Instead of focusing on the functional groups, we’re actually doing the chemistry on the C–H bonds themselves and, in principle, we’re doing it even in the presence of what are considered to be the reactive groups,’ explains Huw Davies, an organic chemistry professor at Emory University, Atlanta, and director of a US-wide collaboration called the Center for Selective C–H Functionalization. ‘That gives us a totally different way of putting molecules together. It often allows you to get into new chemical space, which can be very useful for areas such as new drug development, agrochemicals and material science.’
As well as allowing chemists to make molecules they couldn’t previously, targeting C–H bonds in this way can also shorten and simplify synthetic routes to those that can already be reached. The ability to add the functionality required in the final molecule, such as an oxygen or nitrogen atom, towards the end of its synthesis rather than early on is one of the significant advantages offered by C–H functionalisation.
How do you get the chemistry to work so you can target a specific C–H bond in a practical manner?
Huw Davies
‘Classical organic synthesis focuses on building the hydrocarbon scaffolds while concurrently introducing the oxygen and nitrogen. Usually the two are linked, with things like aldol or Mannich reactions used to build the hydrocarbon scaffolds,’ explains organic chemist Christina White at University of Illinois, US. ‘If you introduce the oxygen or nitrogen as you go along, you have to protect it, deprotect it, manipulate its oxidation state. This leads to very lengthy chemical manipulation. Being able to [add oxygen or nitrogen at a late stage] allows you to really streamline these syntheses, by minimising functional group manipulation and oxidation state changes.’
Another related advantage offered by C–H functionalisation, and which is of particular interest to the pharmaceutical industry, is the ability to make derivatives of a molecule in a single step. ‘Nature oftentimes will build one complex scaffold and then introduce different oxidised functionality, making a lot of different derivatives of a molecule really efficiently. Classically an organic chemist who wanted to make eight different derivatives of, let’s say, Taxol with new functionality in new positions would have to do eight de novo syntheses,’ explains White. C–H functionalisation techniques have now been used to derivatise a range of different natural products at the end of their synthesis, a technique known as late-stage C–H functionalisation.1 Nature uses enzymes to carry out these manipulations, and in turn organic chemists have turned to small molecule metal catalysts for all areas of C–H functionalisation.
Site selectivity
Using a range of different metal catalysts, it is now possible to convert C–H bonds into numerous other bond types including carbon–carbon, carbon–oxygen, carbon–nitrogen and carbon–halogen. And they are able to pluck hydrogen off carbon atoms in both aromatic and aliphatic compounds. But maybe most impressively of all they are now starting to have good control over which of the many C–H bonds in a molecule it is that reacts.
‘The challenge is, when every molecule has lots of C–H bonds, how do you get the chemistry to work so you can functionalise a specific C–H bond in a practical manner,’ explains Davies. Sometimes the answer is to borrow the directing group concept from classical organic synthesis. ‘Using a directing group, which can coordinate to the metal catalyst, places the metal close to a specific C–H bond and allows a selective functionalisation to occur.’
And while normally the directing group and the C–H bond in question are just a few atoms away, they are not always. In 2016, Melanie Sanford at the University of Michigan, US, reported a method for inserting carbons contained within aromatic rings into remote C–H bonds in cyclic aliphatic amines. Here, the nitrogen of the amine acts as the directing group.2
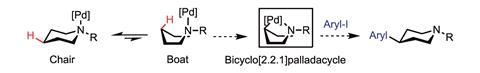
Her team first attached a palladium catalyst and a coordinating group to the nitrogen in the ring. When the ring then naturally flipped from the chair to the boat conformation, the palladium inserts into a C–H bond on the opposite side of the ring, fixing the ring in this less-favourable position. At this point, the palladium is strongly coordinated to both the nitrogen of the amine and the carbon of the original C–H bond. The final step is to add an aryl halide, forming a new C–C bond and releasing the palladium catalyst. ‘We really liked the chemical challenge of designing a catalytic system that would allow you to selectively functionalise far from nitrogen,’ Sanford says.
A number of new chiral ligands with much broader scope are also on their way
Jin-Quan Yu
Meanwhile, Jin-Quan Yu at Scripps Research Institute in California, US, is developing an alternative approach that utilises weaker coordinations between the substrate and catalyst. Yu has demonstrated the utility of his approach for C–H functionalisations at remote sites, but also – most significantly – he has been able to incorporate chiral ligands, allowing the C–H functionalisation to be enantioselective.
‘If we want to develop enantioselective C–H activation reactions in the context of asymmetric catalysis, previous reactions driven by strong coordination with substrates are essentially background reactions that could prevent enantiocontrol,’ says Yu. ‘Therefore, it is critical to use weakly coordinating substrates that are not reactive under ligandless conditions, and ensure that the reactivity is driven by ligand acceleration thereby achieving enantioselectivity.’
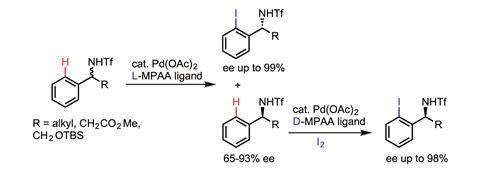
So far, Yu’s first chiral ligand (a mono-protected amino acid) has been successfully applied to develop a ‘diverse range of enantioselective C–H activation reactions’, he adds.3 In these reactions, C–H bonds are converted to a wide range of carbon–carbon and carbon–heteroatom bonds, and the chirality is amplified by either desymmetrisation or kinetic resolution depending on the structure of the substrates. ‘A number of new chiral ligands for enantioselective methylene C–H activation with much broader scope are also on their way.’
Coordination not required
Coordination is not the only approach for controlling site selectivity. White and Davies instead favour an alternative technique known as catalyst-controlled C–H functionalisation. ‘The advantage here is that you don’t need to add the directing group because the controlling element is going to come from the catalyst,’ explains Davies. Here, the catalyst properties are tuned to control which C–H in a molecule will react. This is possible because these bonds all have subtly different electronic and steric properties that the right catalyst is able to pick out.
In 2007 and 2013, White reported two small molecule iron catalysts – Fe(PDP) and Fe(CF3PDP) – that can form carbon–oxygen bonds in aliphatic molecules in a stereospecific and site-selective manner without the use of directing groups.1,4 ‘This work demonstrated that the site of reactivity can be rationally altered by modifying the catalyst,’ she says.
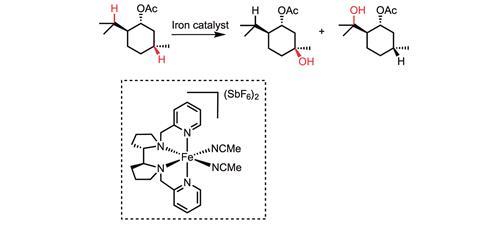
Her group is focused on forming both carbon–oxygen and carbon–nitrogen bonds via the insertion of reactive metal-bound oxo and nitrene species. Here, the hydrogen is first plucked from the molecule by an oxo or nitrene to generate a short-lived carbon-centered radical. This is followed by such rapid Fe–OH functionalisation from the metal to form the hydroxylated product that there is no time for even a bond rotation. This means that if you start with a chiral centre, your products are also chiral.1 ‘Mechanistic studies revealed that both the C–H abstraction step and the functionalisation step are tightly regulated by the metal centre, providing the ability to exert such high levels of catalyst control over reactivity,’ claims White.
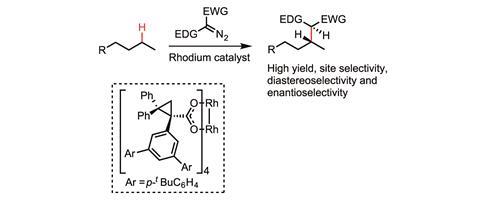
Davies, meanwhile, is focused on forming C–C bonds, by inserting a metal-bound carbenoid into the C–H bond. Donor–acceptor carbenoids are his particular speciality. ‘Conventional carbenoids typically have just acceptor groups on them, making them incredibly electron-deficient, and therefore really reactive and consequently not particularly selective,’ Davies says. ‘We developed these donor–acceptor carbenes where the donor groups significantly stabilise the carbene. We’re able to do [C–H functionalisations] with pretty significant site selectivity. We also can often do it with high enantioselectivity and, in many cases, we generate more than one stereocentre so we can do it with good diastereoselectivity as well.’
In 2016, Davies’ group developed a dirhodium catalyst able to selectively functionalise a C–H bond on the second carbon in pentane in preference to those on the first or third carbon.5 This is significant because there are only really subtle differences between the C–H bonds in this molecule. The reaction was achieved with more than 95% site selectivity and also with good stereocontrol.
Functionalising small hydrocarbons is currently in vogue, with Sanford reporting the C–H borylation of methane in 2016.6 ‘Methane is the primary component of natural gas and ways of selectively turning methane into value-added chemicals are of great interest to the petroleum and natural gas industries,’ explains Sanford. ‘Typically, if you try to functionalise methane it just goes all the way to CO2 – you can’t just functionalise one of the C–H bonds as the product is more reactive than the starting material.’ However, her group was able to do exactly this, inserting boron into just one of methane’s C–H bonds, using a handful of different metal catalysts. ‘The reaction that we have is pretty far from application now – it’s too expensive for example – but there’s a really fundamental interest in being able to control selectivity in methane functionalisation,’ she adds.
Tackling inertia
Carbon–hydrogen bonds are not the only ‘inert’ bonds that chemists are flexing their synthetic muscles trying to break. Neil Garg, for example, is working on turning the strong carbon–nitrogen bond in amides into carbon–carbon bonds. His tool of choice is transition metal catalysed cross-coupling reactions.
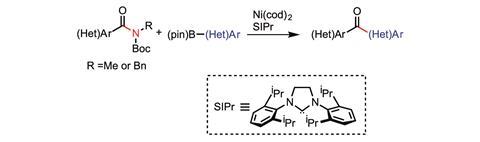
‘In an amide, the carbon–nitrogen bond is significantly strengthening by resonance effects. It’s not easy to break an amide because that C–N bond has a significant amount of double bond character,’ explains Garg. Using a nickel catalyst, his group have successfully swapped C–C bonds for C–N bonds in a range of different amides. This means they have converted an amide to a ketone.
In the reactions, the nickel catalyst is activating the amide’s C–N bond, allowing it to undergo a cross-coupling with a boronic ester to form the C–C bond. Garg’s team have successfully added both aromatic8 and non-aromatic9 carbons in to the C–N bond.
His team has now established proof of concept with this reaction and ‘seen a lot of promise for tolerating things like heterocycles and pretty sterically hindered substrates’, explains Garg. ‘The goal now is to broaden the scope and develop for the chemistry to reach a broader user base.’
Chemoselectivity
Another significant hurdle for all of these different catalytic systems, on which there has been much work, is achieving chemoselectivity. That involves ensuring that the C–H bond reacts with the catalyst in preference to another type of bond in the molecule that would normally be deemed to be more reactive. Some startling examples of this have now been published. In 2015, for example, White reported a manganese catalyst that is able to selectively insert a nitrogen into aliphatic C–H bonds even in the presence of the very reactive C=C double bond.7 This particular reaction is designed to install a nitrogen three atoms away from an oxygen-containing functional group. ‘It takes readily-available natural products that generally are abundant in oxygen functionality and allows you to make 1:3 amino alcohol motif,’ explains White.
The field of C–H functionalisation has exploded in a myriad of different directions in recent years, with many clever reactions having now been developed. But chemists may have only just started to scratch the surface of what can be achieved. ‘The field of C–H activation offers such a fertile ground for discovering new reactions, new catalysis, and the community is far from reaching the full potential of C–H activation,’ says Yu.
Nina Notman is a science writer based in Salisbury, UK
References
1 M S Chen and M C White, Science, 2007, 318, 783 (DOI: 10.1126/science.1148597)
2 J J Topcewski et al, Nature, 2016, 531, 220 (DOI: 10.1038/nature16957)
3 L Chu, K-J Xiao and J-Q Yu, Science, 2014, 346, 451 (DOI: 10.1126/science.1258538)
4 P E Gormisky and M C White, J. Am. Chem. Soc., 2013, 135, 14052 (DOI: 10.1021/ja407388y)
5 K Liao et al, Nature, 2016, 533, 230 (DOI: 10.1038/nature17651)
6 A K Cook et al, Science, 2016, 351, 1421 (DOI: 10.1126/science.aad9289)
7 S M Paradine et al, Nat. Chem., 2015, 7, 987 (DOI: 10.1038/nchem.2366)
8 N A Weires, E L Baker and N K Garg, Nat. Chem., 2016, 8, 75 (DOI: 10.1038/nchem.2388)
9 B J Simmons, ACS Catal. 2016, 6, 3176 (DOI: 10.1021/acscatal.6b00793)










No comments yet