
US organic chemists have devised a powerful pair of synthetic strategies involving nitriles, offering new ways of controlling stereochemistry. Amir Hoveyda from Boston College and his team can now catalytically and enantioselectively convert nitriles to multifunctional amines without any protection, deprotection or oxidation-state adjustments.1 ‘To our surprise, our report is the first example of catalytic enantioselective carbon-based nucleophile additions to nitriles,’ says Hoveyda, who is now setting up a group at the University of Strasbourg, France.
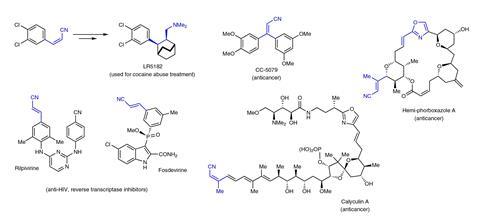
Hoveyda’s team had been trying to improve on the existing 26 step, 10% yield synthesis of potential anticancer drug (+)-tangutorine, originally isolated from Nitraria tangutorum leaves. The chemists wanted to form a fragment with a chiral primary amine group next to another stereogenic centre, requiring careful control over symmetry. However, existing synthetic approaches needed to add a protecting group to the amine, whose removal led to two undesired side-reactions.
To find an alternative that doesn’t need protection, Hoveyda’s team reacted nitriles with monosubstituted allenes and bis(pinacolato)diboron in the presence of a copper complex, making β-boryl ketimines. However, they couldn’t isolate this intermediate – instead the chemists reduced it to the primary amine system with polymethylhydrosiloxane. ‘What is left behind is an alkenyl boronate, one of the most versatile and widely used functional groups in chemistry,’ stresses Hoveyda.
While this successfully gave the primary amine fragment group the scientists wanted, controlling the stereochemical outcome was much harder. A key factor is that the copper complex both helps transfer pinacolatoboron to the ketimine, and then mediates the ketimine’s hydride reduction.

To control rates of both processes, Hoveyda’s team introduced a side reaction cycle to keep the hydride–copper complex occupied until after enough pinacolatoboron–copper complex had formed. This ‘delayed catalysis’ holds the more rapidly generated, more active hydride–copper complex in check until the less active, more slowly produced pinacolatoboron–copper complex has done its job.
Under these conditions, the boron atom holds the ketimine nitrogen in place, ensuring it’s selectively reduced from one face. The chemists then disrupted that symmetry to produce the opposite diastereoisomer, breaking the boron–nitrogen link using a Lewis acid, and adding a chiral ligand. Hoveyda’s team used the approach to prepare tangutorine in just nine steps, in 28% overall yield.
Edward Anderson from the University of Oxford calls the development a ‘very well-constructed piece of research that comprehensively addresses the problem of stereocontrolled amine synthesis from readily available nitriles’.
Bring on the nitriles
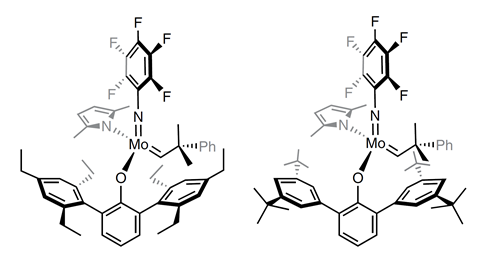
Di- and tri-substituted alkenyl nitriles are potentially powerful starting materials for this strategy, but Hoveyda notes that they were not previously easily accessible. Consequently his team has also developed cross-metathesis strategies, using molybdenum-based monoaryloxide pyrrolide (MAP) or chloride (MAC) complexes, to selectively produce such alkenyl nitriles.2
The approach can work in the presence of functional groups including sulfides, epoxides, alcohols and carboxylic acids elsewhere in the molecules produced. ‘With an appropriate catalyst one can prepare virtually any alkenyl nitrile, E- or Z-, di- or tri-substituted,’ Hoveyda observes.
MAP complexes enable the team to produce Z-di-substituted alkenyl nitriles starting from acrylonitrile and terminal alkenes, although bulky or electron-withdrawing substituents on the alkene reduced reaction yields. The Boston College scientists therefore turned to MAC complexes in these cases. However, because MAC complexes degrade in the presence of terminal alkenes, the chemists had to switch to maleonitrile and Z-alkene starting materials.
Similarly, synthesising E-alkenyl nitriles worked best with fumaronitrile and E-alkene starting materials and a slightly different molybdenum MAC complex. Hoveyda observes that it’s more important to have the flexibility to find the best catalyst for a particular reaction than just one catalyst that might not be suitable for every case. ‘High selectivity and high generality cannot co-exist,’ he stresses.
Anderson says that the team’s development of the cross-metathesis approach is a ‘substantial piece of work’. ‘That this chemistry can access di-substituted and tri-substituted alkenyl nitriles, with exquisite control over alkene geometry, is really impressive,’ he says.
Hoveyda is confident that the strategy will prove important, with the molybdenum metathesis approach already being exploited. ‘These catalysts are being applied in industry to metric ton scale reactions,’ he says. ‘For smaller scale applications, one can purchase these catalysts from well-known vendors as paraffin tablets that are air and moisture stable. Nothing could be easier.’
References
1 S Zhang et al, Science, 2019, 364, 45 (DOI: 10.1126/science.aaw4029)
2 Y Mu et al, Nat. Chem., 2019, DOI: 10.1038/s41557-019-0233-x















No comments yet