A single hydrogen bond can stabilise a transition state significantly more than its intrinsic interaction energy would suggest, new research shows. The team behind the work suggest that stabilisation partly arises from the molecule using strong destabilising repulsive interactions to prepay an energy penalty reaction – a concept that chemists could use to make better catalysts.
Hydrogen bonds can play crucial roles in the kinetics of enzymatic or synthetic organocatalytic systems, but the fleeting nature of transition states means these effects are difficult to measure.
Now, Ken Shimizu’s team at the University of South Carolina has designed a series of molecular rotors to study and measure the kinetic effects of an intramolecular hydrogen bond on the rate of a bond rotation. The rotors in question have an N-phenyl unit attached to a five-membered imide ring by a single carbon–nitrogen bond. They carried out experiments using three separate dynamic 1H NMR methods, which all gave similar results – a significantly lower rotational barrier for rotor 1, which has a hydroxyl group on the N-phenyl unit, than the methoxy group-containing rotor 2.
‘We expected the rotational barrier energy to be low because of the ability of rotor 1 to form a stabilising bond in the transition state,’ says Shimizu. ‘I estimated the strength of that hydrogen bond to be between 1–3kcal/mol based on previous measurements of similar hydrogen bonds. However, we measured barriers that were the result of transition state stabilisations of 9–11kcal/mol, which seemed unbelievable at first.’
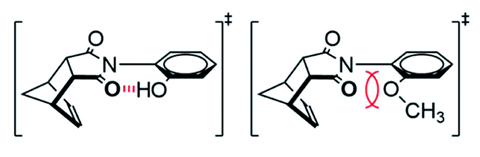
The large stabilisation partly resulted from rotor 1 being designed to minimise the repulsive component of the hydrogen bonding interaction. ‘Most chemists are used to thinking about the energetics of hydrogen bonds in terms of their intuitive attractive components,’ comments Scott Cockroft, whose research at the University of Edinburgh, UK, examines the physical organic chemistry underpinning molecular interactions. ‘This work elegantly reveals the importance of repulsive contributions in constrained intramolecular systems.’
The team reduced the repulsive component of the hydrogen bond by fixing the key interacting groups in close proximity in the hydrogen bonding transition-state structure, which is often the case in enzymes. This means that the energy penalty of the reaction is paid during initial formation of the complex, and there is less of a barrier to overcome during the reaction. ‘These rotor systems are simple and elegant. They mimic the effects of constrained hydrogen bonds in an enzyme active site, but stripped of all the complexity,’ comments Judy Wu, from the University of Houston, US, who uses aromaticity to control non-covalent interactions. ‘We get a glimpse of the upper bound estimate of the stabilisation that non-covalent interactions can contribute to catalysing a reaction – and the magnitude of the effect is quite surprising.’
The findings could explain certain enzyme mechanisms. It could also be used by researchers when designing new hydrogen bonding catalysts. ‘In addition to the potential applications in catalysis, I envisage that such characteristics be could be exploited in molecular devices that switch hydrogen bonds on and off to control nanomechanical conformational changes,’ adds Cockroft.
Shimizu is currently looking further into the fundamentals of the system, and is confident that the molecular rotors will allow this. ‘We’re excited about using this as a tool to interrogate transition states.’











No comments yet