Deep-learning network AlphaFold2 – trained on about 170,000 protein structures – has caused great excitement in the scientific community
A team at Google offshoot DeepMind say that their artificial intelligence (AI) network has made a huge leap solving the 50-year-old protein folding problem after it outcompeted all other teams at in a protein structure prediction challenge. The program has been received with enthusiasm by researchers worldwide who say it could revolutionise biology, in particular areas such as drug design or environmental sustainability.
Fantastic scientific breakthrough. One of the biggest problems in science (protein folding) looks like it’s been cracked by artificial intelligence. Huge implications for medicine. Congratulations @DeepMind #AlphaFold https://t.co/qwbiJINMBh
— Jim Al-Khalili (@jimalkhalili) November 30, 2020
Google’s algorithm, called AlphaFold2, was the clear winner of this year’s Critical Assessment of Structure Prediction (Casp14) challenge – a biennial competition created to benchmark progress in the accuracy of computational protein structure prediction. AlphaFold2 was able to determine the 3D shapes of around two thirds of target proteins with an accuracy comparable to laboratory experiments, greatly outperforming around 100 other teams.
Proteins are complex molecules comprised of amino acid chains that can fold into different shapes. Predicting what the final 3D structure will look like is a challenge that scientists have worked on for decades. Mohammed AlQuraishi from Columbia University, US, who developed one of the competing algorithms, thinks that DeepMind’s AI could help researchers understand the functions of proteins by comparing their predicted structures to proteins with known function and shape. He adds that although the program wasn’t developed for protein design, experience from other models like Rosetta suggests that it might be possible to adapt it for this purpose. ‘It’s likely not yet accurate enough for medical applications focused on small molecules, but it may help with designing antibodies for protein-based therapeutics,’ he says.
Andrei Lupas at the Max Planck Institute for Developmental Biology in Germany, who was one of the judges at Casp14, believes that there’s a lot of algorithmic wizardry behind the scenes. ‘They haven’t been too clear about what they’re doing, but they’re spending a lot of time getting the details right, and this seems to add up to a very, very good overall prediction,’ he notes. ‘My department provided a target for a protein that we hadn’t been able to solve for a decade. They gave us a model with which we solved the structure in half an hour!’
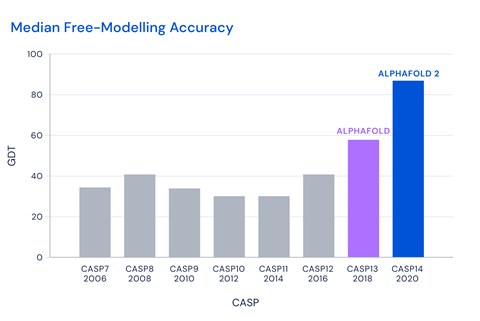
The Casp competition uses the global distance test (GDT) metric to assess accuracy. Any programs reaching a score of around 90 GDT are considered to be competitive with experimental methods. AlphaFold2 achieved a median score of 92.4 GDT across all targets, leaving all its competitors far behind. The software, which was trained on around 170,000 structures from the protein data bank, builds on a previous version that was presented at Casp13 in 2018.
There’s a key difference between the two versions, explains AlQuraishi. ‘This one is end-to-end differentiable, which means the system is optimised to go from sequence to the final 3D structure, and all the pieces in the system are jointly optimised to learn from data,’ he says. ‘The original AlphaFold had multiple separate pieces trained independently and only predicted interatomic distances – not 3D structures – that were used to fold the protein using more conventional approaches like Rosetta.’ AlphaFold2 is iterative, generating an initial 3D structure that is then refined over many steps, ‘so it’s able to extract more complex patterns from the data’, he explains.
Although AlQuraishi’s own model didn’t perform as well as AlphaFold2, it improved over the previous version too. ‘Our method works from individual protein sequences, not from homologous protein sequences like AlphaFold2. We think this route is worthwhile because it may enable the design of proteins very different from naturally occurring ones and may be more sensitive to changes in individual sequences, for example, mutations.’
DeepMind now wants to improve the algorithm further to make it figure out how proteins form complexes or how they interact with small molecules.










No comments yet