The need to use wavefunction or density functional theory (DFT) calculations to determine electron densities has been bypassed by a machine learning model. It will allow chemists to quickly determine properties that depend on the electron density of large systems such as van der Waals forces, halogen bonding and C-H–π interactions. These non-covalent interactions can hold insight into the binding of host–guest systems or favoured enantiomers within reaction pathways where intermediates and transition states may be stabilised by subtle attractions.
The electron density distribution is one of the most powerful tools at the disposal of a computational chemist. From the electron density, properties such as charges, dipoles and electrostatic interaction energies can be determined. Accurately accounting for these is essential for the predictive power of many quantum chemistry techniques such as computing infrared intensities or determining non-covalent interactions.
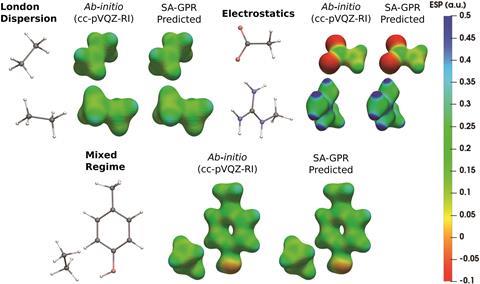
Computing the electron density can be challenging and time consuming for large systems using traditional wavefunction or DFT methods. To overcome this issue, Clémence Corminboeuf, Michele Ceriotti and colleagues at the Swiss Federal Institute of Technology (EPFL) have developed a machine learning model that can predict the electron density from only atomic coordinates. ‘The breakthrough is to be able to accurately predict, in a few minutes at most, the electron density of complex molecules without any quantum chemical computation,’ explains team member Alberto Fabrizio.
‘I think it’s a really interesting approach, both in terms of prediction errors and transferability to small and large systems,’ comments Natalie Fey who researches computational inorganic chemistry at the University of Bristol, UK.
Quantum tricks for better machine learning
The machine learning model relies upon a vast training set of small molecule dimers. These dimers have their electron density represented by basis sets, similar to those used in normal computational chemistry calculations. The trick behind increasing the accuracy of the predicted electron density was to use auxiliary basis sets that are designed for the resolution-of-the-identity method – an approximation that helps speed up calculation of two electron integrals. ‘The key advancement in this work, compared to their previous work, is that the researchers have now introduced auxiliary functions to efficiently parametrise the densities to be predicted,’ remarks Markus Reiher, a theoretical chemist at the Swiss Federal Institute of Technology in Zurich.
‘Standard basis sets are constructed to approximate as closely as possible the wavefunction, while auxiliary basis sets within the resolution-of-the-identity approximation are designed to mimic one-electron densities,’ explains Corminboeuf. By using these auxiliary basis sets, the errors in the predicted densities are diminished to beneath 0.5%, far more accurate than the >10% error with more commonly used basis sets such as cc-pVDZ.
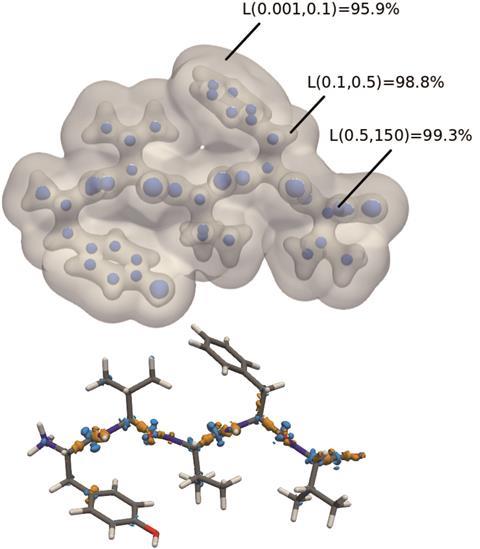
Once the machine learning code had been trained on density functional computed electron densities of the small dimers, the ability to accurately predict electron densities of chemically different, and far larger, systems was probed. The electron density of the 77 atom polypeptide enkephalin was successfully predicted to within 1.4% against DFT calculations and within a series of eight large polypeptides, the average error was only 1.5%. This electron density was achieved far quicker than standard DFT methods, giving quicker access to the all-important electron density and further insights into non-covalent interactions in these large polypeptides.
However, Fey notes that the ‘near-sightedness’ of the current model remains an issue. This refers to the machine learning model representing molecules as 4Å wide atom-centred environments. Due to this, the long-range interactions are not fully captured.
Corminboeuf suggests a possible solution noting that ‘this issue can be tackled by modifying the way we represent our local chemical environments. To encode the non-local information, one could formulate the framework to represent the environments in the form of a potential, integrated over all space.’ For now, the newly developed machine learning model will allow for a host of rapid predictive possibilities so long as atomic coordinates are to hand, whether they come from x-ray crystallography or classical molecular dynamics simulations.










No comments yet